
Abstract
Abstract
Passive biological systems such as sulfate-reducing biochemical reactors have shown promise for treatment of mine drainage because of their low cost, minimal maintenance, and constructability in remote locations. However, few criteria exist for their design and operation. In particular, the impact of the choice of carbon substrate is poorly understood. This study represents the first to directly compare the effect of simple and complex organic substrate on microbial communities present in pilot-scale biochemical reactors treating mine drainage. Three organic substrates were evaluated: ethanol (ETOH), hay and pine wood chips (HYWD), and corn stover and pine wood chips (CSWD). Microbial community compositions were characterized by cloning and sequencing of 16S rRNA and apsA genes corresponding to the sulfur cycle. Quantitative polymerase chain reaction was applied to quantify Desulfovibrio-Desulfomicrobium spp. and methanogens. Results revealed differences in microbial compositions and relative quantities of total and sulfate-reducing bacteria among reactors. Notably, the greatest proportion of sulfate-reducing bacteria was observed in the ETOH reactors. HYWD and CSWD reactors contained similar bacterial communities, which were highly complex in composition relative to the ETOH reactors. Methanogens were found to be present in all reactors at low levels and were highest in the lignocellulose-based reactors. Interestingly, higher proportions of aerobic Thiobacillus spp. were detected in two reactors that experienced an oxygen exposure during operation. This study demonstrates that both substrate and environmental stress influence both microbial community composition and diversity in biochemical reactors treating mine drainage. While there were no significant differences in performance observed over the time scale of this study, potential long-term implications of the differing microbial communities on performance are discussed.
Introduction
A variety of abiotic and biological remediation systems have been developed and used for the treatment of MIW. Abiotic methods usually involve the addition of neutralizing agents or other chemicals and, although effective, they may require impractically high levels of cost, operation, and maintenance (Johnson and Hallberg, 2005a; Cohen, 2006). Alternatively, bioremediation technologies have shown promise because of their low operational costs and minimal external energy requirements. Sulfate-reducing biochemical reactors, such as those applied at the Wheal Jane (Whitehead and Prior, 2005) and Nickel Rim (Benner et al., 1997) mines, have proven successful. However, reports from the field are variable in terms of the overall performance and reliability of these systems (Benner et al., 1997; Barton and Karathanasis, 1999; Johnson and Hallberg, 2002). The basic principle of biochemical reactors is the cultivation of sulfate-reducing bacteria (SRB) that reduce the sulfate in the MIW to sulfide, which in turn reacts with metals and precipitates them as metal sulfides. Metal sulfides are immobilized within the reactor and thus removed from the water (Tuttle et al., 1969; Johnson and Hallberg, 2005b). Significant levels of alkalinity are also microbially produced as bicarbonate, which decreases the acidity level within the reactor.
A significant aspect of biochemical reactor design is the choice of organic substrate (Waybrant et al., 1998; Daubert and Brennan, 2007). Relatively simple substrates such as organic acids or alcohols have the advantage of providing a carbon and energy source that SRB can directly utilize for growth (Tsukamoto and Miller, 1999). In particular, the low freezing point characteristic of alcohol substrates makes passive delivery possible during winter months. The disadvantage, though, is that simple substrates are used quickly and need to be continuously fed into the system, which increases the cost and maintenance (Cohen, 2006). Simple substrates can also be used by a wide variety of species, such as methanogens or acetogens, which may out-compete the desired SRB (Koschorreck et al., 2010). Alternatively, complex lignocellulose-based organic materials (e.g., wood chips) are biodegraded slowly, providing a long-term source of carbon and energy. In this case, SRB rely on the activities of anaerobic cellulose degraders and fermenters to break down the complex material into simpler molecules that support their growth (Logan et al., 2005).
Recent studies using molecular biological tools have revealed the complexity of microbial communities in lignocellulose-based bioreactors (Johnson and Hallberg, 2005b; Pruden et al., 2007; Hiibel et al., 2008), and MIW remediation based on various types of organic substrates have been evaluated (Bechard et al., 1994; Waybrant et al., 1998; Figueroa et al., 2004) and recently reviewed by Neculita et al. (2007). However, direct comparison of the microbial communities resulting from complex versus simple carbon substrates has not been performed. Differences in the recalcitrance of the substrate in biochemical reactors are likely to drive the development of distinct microbial communities in terms of types of microorganisms present, their relative abundance, and overall diversity. This, in turn, can have an impact on performance since microbes are the primary catalysts of bioreactor function. Prior studies have demonstrated that the composition of the microbial community impacts reactor start-up time (Christensen et al., 1996), sulfate reduction rate (Pruden et al., 2007; Pereyra et al., 2008), and metal removal (Weber et al., 2010).
The purpose of this study was to explore the effect of the organic substrate on the resulting microbial communities in parallel biochemical reactors treating MIW. Six pilot-scale reactors were operated in Black Hawk, CO, and were fed with discharge from the National Tunnel, which is a major contributor of contaminants to Clear Creek (Buccambuso et al., 2007). Three types of organic substrate were compared in duplicate: ethanol (ETOH 1,2), hay and pine wood chips (HYWD 3,4), and corn stover and pine wood chips (CSWD 5,6). Biomolecular characterization of the microbial populations included cloning of 16S rRNA and the adenosine-5′-phosphosulfate reductase (apsA) genes, which encodes the catalysis of the first step in the sulfate reduction pathway. Quantitative polymerase chain reaction (Q-PCR) was employed to provide sensitive quantification of total bacteria, the Desulfovibrio-Desulfomicrobium genera of SRB frequently observed in MIW remediation sites, and methanogens. Clearer insight into substrate–microbe interactions can improve understanding of the fundamentals of biochemical reactor function and therefore help develop better criteria for their design, operation, and overall performance.
Experimental Protocols
Sample site and collection
Six pilot-scale biochemical reactors were installed under the Mill Street Bridge in Black Hawk, Colorado, on June 28–29, 2006 (Buccambuso et al., 2007) and fed with a water discharge from the National Tunnel. As a result of historic gold mining, the discharge (pH 6.2–6.5) contains elevated levels of sulfate (250–300 mg/L as S) and heavy metals (5–7 mg/L zinc, 35–50 mg/L iron, ∼0.1 mg/L copper, and 5–7 mg/L manganese). The bioreactors consisted of duplicate 208-L (55-gallon) plastic drums utilizing one of three different organic substrates. The three substrates evaluated were chosen as representative of materials commonly used in MIW remediation, and include (1) ethanol as a simple organic molecule (Tsukamoto and Miller, 1999; Tsukamoto et al., 2004), (2) corn stover and pine wood chips as a complex carbon source with a high cellulose content, and (3) hay and pine wood chips as an additional complex carbon source with higher nitrogen and protein content (Figueroa et al., 2004). The ethanol-fed bioreactors (ETOH 1,2) were packed with limestone with a zero valence iron slag layer on the top (to provide physical attachment sites using an element well-represented in the MIW) and inoculated with ∼2.3 kg of horse manure layered on top of the limestone; the reactors received 100 mL/day of 50% ethanol (0.11% to 0.29% by volume). The lignocellulose-based bioreactors contained a mixture of wood chips, limestone, horse manure (as inoculum), and hay (HYWD 3,4) or corn stover (CSWD 5,6) in the proportions presented in Table 1. Each non-ethanol drum had ∼170 L of reactive media. Note that the densities of the dry materials varied slightly; thus, it was necessary to make minor volume adjustments with the limestone. Limestone was considered to be relatively inert, since it is naturally in equilibrium with water and the pH was equivalent across the reactors. All reactors received ∼34 L per day in six daily 5.7-L increments at the top (down flow reactors). The pore volume and hydraulic residence times were estimated previously to be a 0.3–0.5 v/v and 2.2–3.7 days, respectively (Venot et al., 2008).
HYWD, hay and pine wood chips; CSWD, corn stover and pine wood chips.
During reactor setup, five removable mesh bags (5×10 cm) were filled with the respective substrate and stacked in the center of each reactor within a vertical perforated PVC pipe to facilitate representative sampling across the middle portions of the reactors without disturbing operation. For sample collection, one mesh bag from each reactor (second from the topmost) was removed and placed on ice for immediate transport to the laboratory, where they were stored at −80°C for biomolecular analyses. Sampling was performed on May 16, 2007, after 1 year of operation and during a period of stable sulfate removal, to allow the microbial community to develop and adapt to the biochemical reactor conditions and to approximate steady-state operation.
DNA extraction
DNA was extracted from ∼5 g of each of the collected samples using the PowerMax Soil DNA Isolation Kit (MO BIO, Carlsbad, CA) according to the manufacturer's protocol, and stored at −80°C. The exact mass of material used in each extraction was determined and recorded for downstream quantification purposes.
16S rRNA and apsA gene PCR amplification and cloning
To characterize the overall composition of the bacterial community and to identify dominant bacteria, cloning of 16S rRNA genes was performed. To specifically target SRB, cloning of the apsA gene, which catalyzes the reversible reduction of adenosine 5′-phosphosulfate to sulfite, was also performed. The 16S rRNA gene and apsA gene (Friedrich, 1998) were amplified using primer sets 341F and 1492R [primer 1 of (Muyzer et al., 1993; Weisburg et al., 1991)], and 7F and 8R (Friedrich, 1998), respectively. PCR products were cloned with the TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Inserts were amplified using the vector-specific primers T7 and T3. Restriction-fragment length polymorphism (RFLP), a well-established, technologically simple, and cost-effective technique for screening large clone libraries, was performed from PCR-amplified inserts digested with the MspI restriction enzyme (Promega, Madison, WI). Digest products were separated on agarose gels and a molecular marker was loaded with every eight to ten clones. A high-resolution image of each gel was captured using a UVP BioChemi gel documation system (Upland, CA), and unique RFLP patterns were identified by visual comparison to the markers. The Shannon Diversity Index (H) was calculated as (Cox, 1972):
where S is the size of the clone library and pi is the relative abundance of each restriction pattern. The index provides a comparison between libraries by taking into account both the abundance (richness) and proportion (evenness) of species within a library.
The 16S rRNA gene clones with RFLP patterns occurring two or more times and all apsA gene clones with unique RFLP patterns were sequenced. Because of the labor and cost associated with cloning of the 16S rRNA genes, cloning of these genes was performed on only one of each of the duplicate reactors.
Sequence analyses
DNA sequences obtained from cloning were aligned to the sequences of the most highly similar microorganisms identified by the Basic Local Alignment Search Tool (BLAST) (www.ncbi.nlm.nih.gov/BLAST) and the Ribosomal Database Project II (RDP) (http://rdp.cme.msu.edu). A literature review was performed to assign a putative function to the identified microorganisms. Phylogenetic trees were constructed using the gene sequences of each of the unique clones identified along with sequences of related microorganisms obtained from the National Center for Biotechnology Information Database using the neighbor-joining method with the Jukes and Cantor correction algorithm in PAUP version 4.0b10 (Swofford, 2002). All sequences were deposited to GenBank under accession numbers HM460893 to HM460975 (16S rRNA genes) and HM460976 to HM461041 (apsA genes).
Quantitative polymerase chain reaction
All reactions were performed on a 7300 Real Time PCR cycler (Applied Biosystems, Foster City, CA). Samples were analyzed in triplicate using 12.5 μL 1×Power SYBR Green PCR Master Mix (Applied Biosystems), 0.3 μM of primers DSV230f/DSV838r (Daly et al., 2000), and 0.25 mM of Mg(OAc)2 for the Desulfovibrio-Desulfomicrobium Q-PCR or 0.2 μM of mcrA-targeted primers (Luton et al., 2002; Pereyra et al., 2010) and 1 mM of Mg(OAc)2 for the methanogen Q-PCR. In addition, 1 μL of template DNA and deionized water to a final volume of 25 μL were added to the master mix. The temperature programs included 10 min at 95°C followed by 60 cycles of 95°C for 30 s, 64°C for 37 s, and 72°C for 60 s for the DSV primers or 50 cycles of 95°C for 30 s, 56°C for 40 s, and 72°C for 40 s for the mcrA (methyl coenzyme-M reductase) primers. Total bacterial 16S rRNA genes were quantified using the conditions, universal BACT1369F and PROK1492R primers, and the TM1389F probe described by Suzuki et al. (2000).
For the total bacteria and Desulfovibrio-Desulfomicrobium Q-PCRs, a 5 to 6-point calibration curve was constructed using purified PCR product amplified with primers 8F and 1492R (Eden et al., 1991; Weisburg et al., 1991) from a sample of the CSWD 6 bioreactor and from a Desulfovibrio salexigens pure culture as standards, respectively. Genomic DNA from Methanococcus maripaludis (ATCC 43000D) served as a standard for methanogens. Standards were analyzed in triplicate. The calibration curves were used for absolute quantification of the target genes under the assumption that the amplification efficiency of standards and samples was the same. To ensure that only samples for which this assumption was valid were analyzed, the procedure proposed by Chervoneva et al. (2006) was applied on the Q-PCR amplification efficiencies calculated with LinRegPCR (Ramakers et al., 2003).
Statistical analyses
The phylogenetic trees of the 16S rRNA and apsA genes were analyzed directly using principal coordinate analysis (PCoA) and the UniFrac significance tests of the UniFrac algorithm (Lozupone and Knight, 2005). PCoA differs from principal component analysis in that the input data are the phylogenetic distances between each pair of sampling locations rather than the number of times each clone sequence was observed at each location. The UniFrac significance test is used to determine whether two microbial communities are significantly different (Lozupone and Knight, 2005). For all analyses, non-normalized abundance data, based on the RFLP screening, were incorporated in the UniFrac significance test. Reference strains used to construct the phylogenetic trees were not included in the analysis. A p-value <0.05 was considered to be statistically significant.
Q-PCR data were analyzed using the PROC GLM function of SAS 9.1 (SAS Institute Inc., Cary, NC). Significance was defined by a pair-wise comparison p-value ≤0.05.
Results
Biochemical reactor performance
Performance of the bioreactors has been the subject of prior studies (Buccambuso et al., 2007; Venot et al., 2008). In brief, the performance of all reactors was very similar over the 1-year study period. All reactors achieved a slight increase in effluent pH (6.2–6.5) with respect to the influent. After the first month of operation, effluent Eh of all reactors averaged −230±40 mV, indicative of a reducing environment. All reactors readily removed zinc to below 0.1 mg/L after the first month. Sulfate reduction started peaking in the second and third months, with effluent sulfate concentrations between 50 and 90 mg/L as S. Sulfate removal was highly variable, but was typically about 75%. In the colder months, it appeared that sulfate reduction was slightly higher for ethanol fed reactors (Buccambuso et al., 2007). During the ∼1 year of operation, ambient temperatures ranged from −13°C (December) to 36°C (July), which likely had a stronger impact than is typical because the reactors were maintained above ground. Data were sporadic after 6 months due to heavy snow and freezing conditions at the site (the area experienced a blizzard December 20–21, 2006). At one point, damage to the HYWD 3 reactor due to freezing lead to extensive leakage and exposure of the substrate to the atmosphere that could not be repaired for several weeks because of lack of access to the site. The ETOH 1 reactor experienced some atmospheric exposure as well, but not as extensive, and both bioreactors were repaired and operations continued until samples were collected in May 2007. At the time of this study, all reactors had recovered and were achieving stable sulfate reduction.
Microbial community diversity and composition
Rarefaction analysis of RFLP patterns revealed that the overall bacterial diversity of the lignocellulose-based CSWD 5 bioreactor (H=2.97) was significantly higher than that of the ethanol-fed ETOH 2 bioreactor (H=1.03) (Fig. 1). The overall diversity of the HYWD 3 reactor (H=2.05), which had been exposed to the atmosphere, was intermediate compared to the other two reactors (Fig. 1).
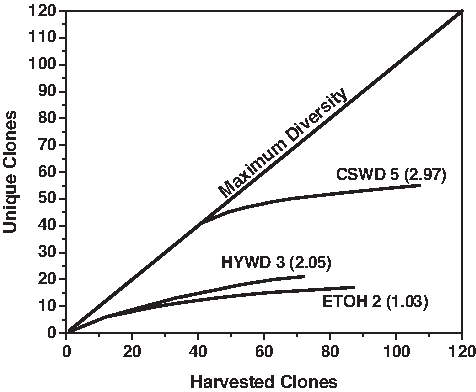
Relative diversity of ETOH 2, HYWD 3, and CSWD 5 biochemical reactors determined by cloning of the 16S rRNA gene. Rarefaction curves were created by plotting the number of unique species identified versus the number of individuals sampled. Shannon diversity index for each reactor is indicated in parenthesis and was calculated based on the frequency of each clone in the clone library. ETOH, ethanol; HYWD, hay and pine wood chips; CSWD, corn stover and pine wood chips.
Sequencing the 16S rRNA gene of the dominant clones also revealed differences between the reactors. Notably, the proportion of SRB in the ETOH 2 reactor was almost 70%, whereas SRB were only 2%–5% in the HYWD 3 and CSWD 5 reactors of the clones identified from the 16S rRNA gene sequencing (Fig. 2). Other putative functional groups, such as cellulose degrading bacteria, were identified in the lignocellulose-based reactors but not in the ethanol reactors, whereas fermentative bacteria were estimated to be 25%–30% of the lignocellulose-based communities and only 5% of the ethanol-based community, based on 16S rRNA gene sequences. Only one putative acetogen was detected, and it was found in the ETOH 2 reactor (data not shown).
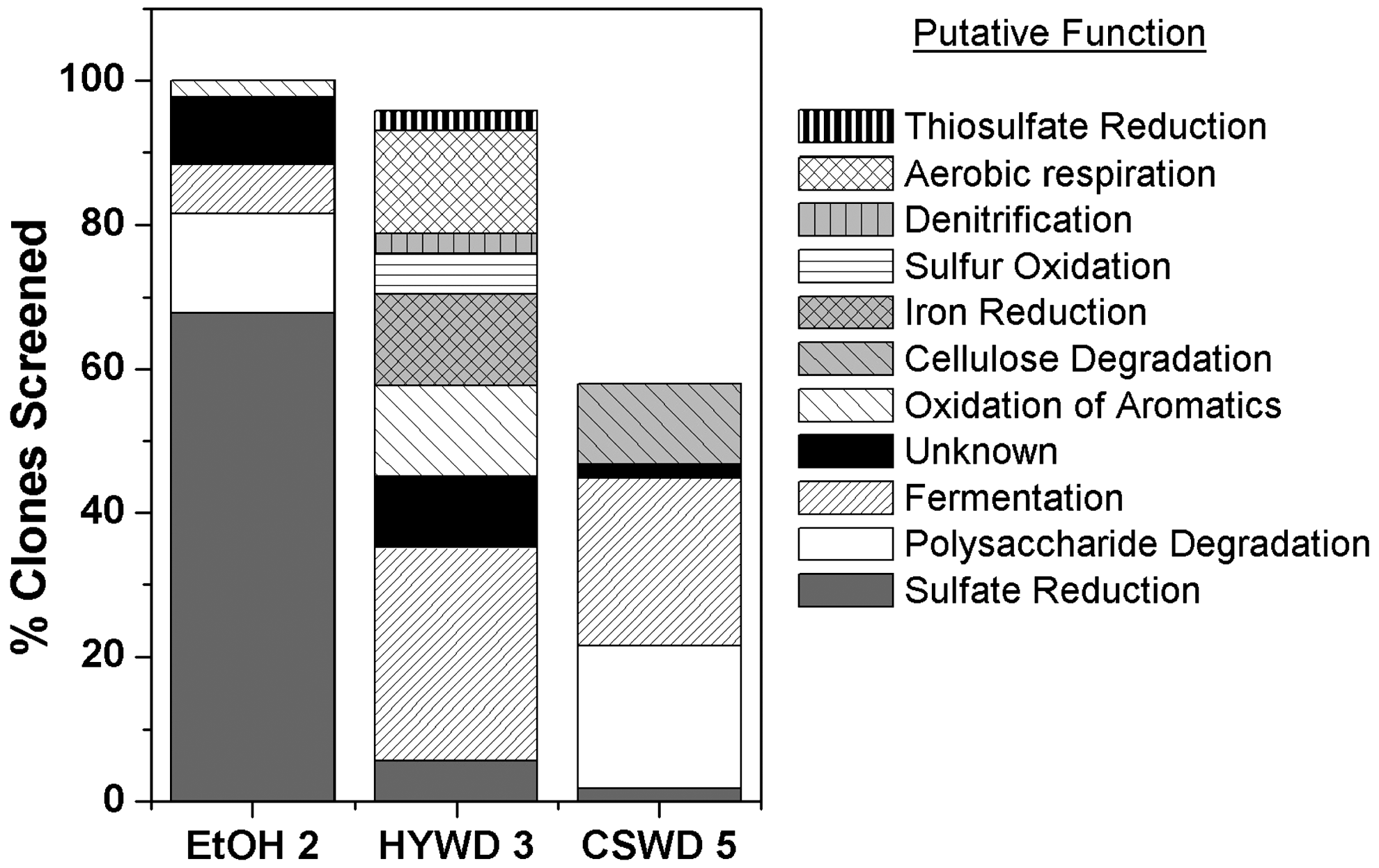
Distribution of putative microbial functions in each biochemical reactor based on the identity of the microorganisms identified through 16S rRNA gene cloning and DNA sequencing of clones. Over 100 clones were evaluated for each bioreactor (101 for ETOH 2 and CSWD 5; 102 for HYWD 3). Low frequency clones (appearing only one time in the library) were not sequenced, accounting for the difference between the % clones screened and 100%. The polysaccharide degradation category (white) includes all non-cellulose polysaccharides.
The apsA gene profiling revealed a low level of SRB diversity, only three to four species, in all of the reactors (Fig. 3). An SRB most closely related to Desulfovibrio sp. JD160 was the only one found in all of the reactors, though notably higher in lignocellulose-based bioreactors that were not exposed to the atmosphere (i.e., HYWD 4, CSWD 5, and CSWD 6). At the same time, there appeared to be a general dominance across the reactors of apsA genes with high sequence similarity to SRB known to tolerate oxygen. Notably, an apsA gene sequence highly similar to that of Desulfovibrio aerotolerans (Mogensen et al., 2005) was found in four of the six reactors. Desulfovibrio desulfuricans has similarly been reported to tolerate oxygen (Cypionka, 2000), but a corresponding apsA gene sequence was only detected in the CSWD 6 reactor and at a relatively small proportion. By contrast, the two SRB detected that have been reported to be highly sensitive to oxygen, Desulfovibrio burkinensis (Ouattara et al., 1999) and Desulfomicrobium baculatum (Copeland et al., 2009), were found collectively in only three reactors and represented a much smaller fraction of the total apsA gene pool compared to D. aerotolerans. Interestingly, one of the reactors in which D. baculatum was detected, HYWD 3, had previously experienced aerobic stress, suggesting that anaerobic conditions had been re-established in this reactors at the time of the study. On the other hand, the dominance of Thiobacillus spp. in HYWD 3 served as a strong indicator of prior exposure of this reactor to the atmosphere. Thiobacillus spp. have been recognized in the literature as aerobic and denitrifying organisms that reverse the process of sulfate reduction (Baker and Banfield, 2003), which corresponds to the atmospheric exposure this reactor underwent. It should be noted that because apsA is subject to horizontal gene transfer (Friedrich, 2002), species assignments are not precise, but do provide a good approximation of the kinds of SRB present.
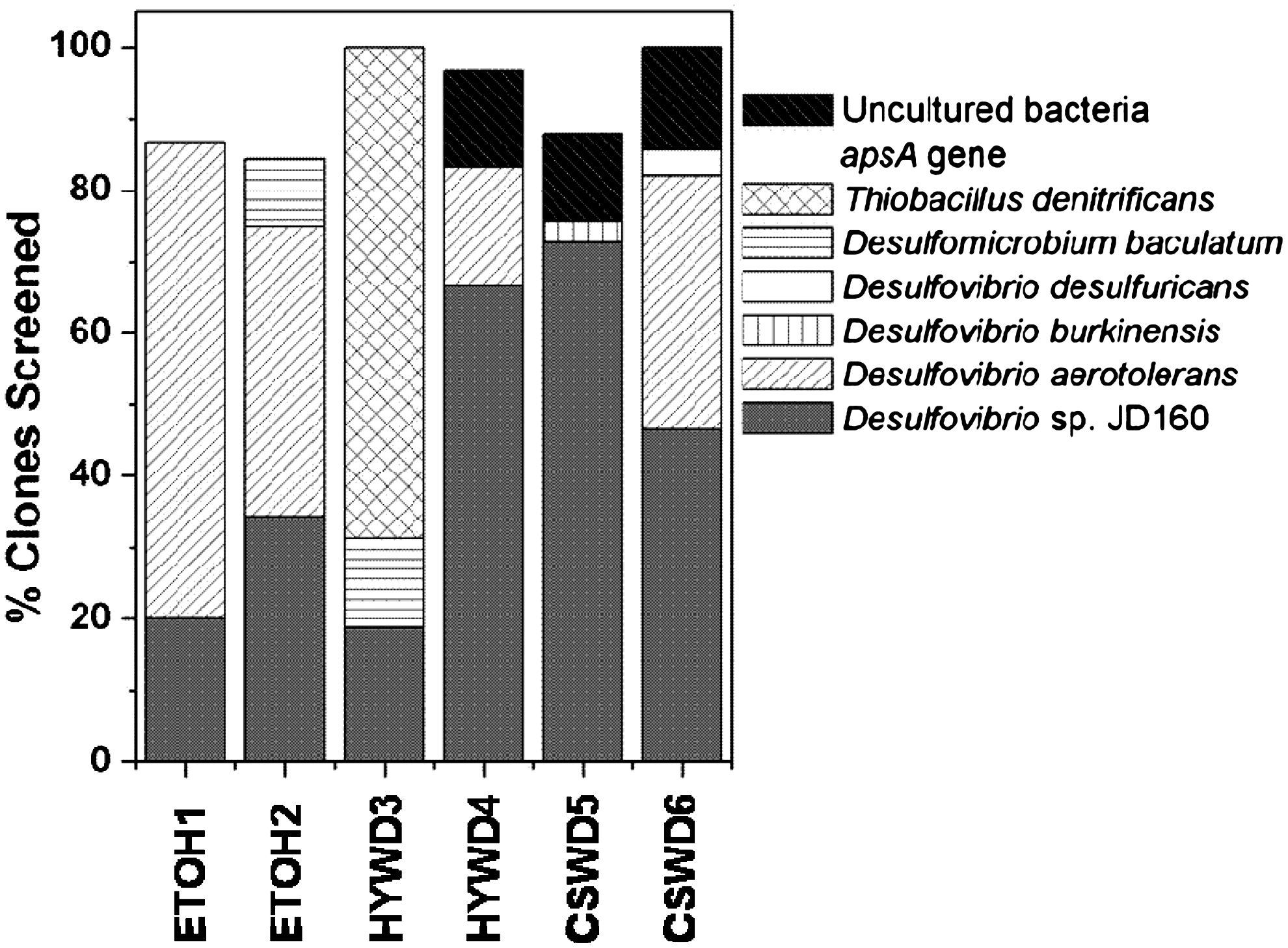
Identity and percentages of apsA-containing microorganisms in ETOH, HYWD, and CSWD biochemical reactors. Approximately 30 clones were evaluated for each bioreactor (29 for ETOH 1, 32 for ETOH 2 and HYWD 3, 30 for HYWD 4, 33 for CSWD 5, and 28 for CSWD 6). Low frequency clones (appearing only one time in the library) were not sequenced, accounting for the difference between the % clones screened and 100%.
PCoA provided a means to compare the microbial communities statistically (Fig. 4). The resulting clustering along each of the two axes serves as a visual representation of the relative similarity of the microbial communities as determined by the DNA sequences and their relative abundances estimated by RFLP. PCoA analysis of 16S rRNA genes indicated that the ETOH 2, HYWD 3, and CSWD 5 communities were distinct across both coordinates. PCoA analysis of apsA genes, however, demonstrated that all of the reactors except HYWD 3 were relatively similar along the first coordinate, which explained 73% of the variability observed. There was more distinction noted among the reactors along the second coordinate, with some modest grouping between duplicates (except for those experiencing known operational problems). Interestingly, an examination of the apsA PCoA data points corresponding only to those for which 16S rRNA gene sequences are available reveals a similar dispersed pattern, with HYWD 3 distinct and CSWD 5 and ETOH 2 most similar along the first coordinate.
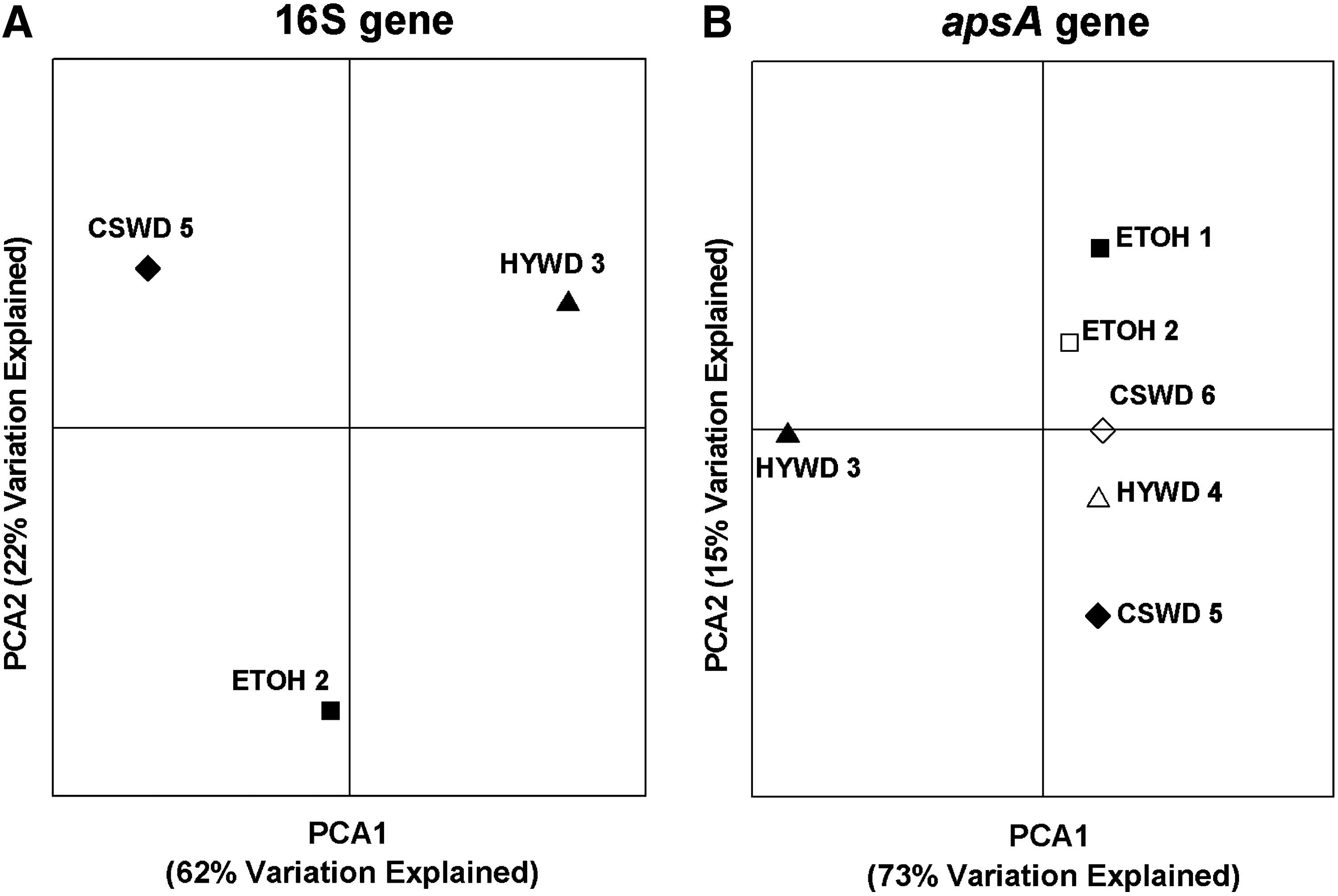
Principal coordinate analyses (PCoA) of the
Quantification of total bacteria, SRB, and methanogens
The results from the total bacterial, Desulfovibrio-Desulfomicrobium, and mcrA Q-PCR were used to estimate the size of the bacterial communities in each reactor (expressed as number of 16S rRNA gene copies per mass of substrate) (Fig. 5A). 16S rRNA genes corresponding to Desulfovibrio-Desulfomicrobium were then normalized to the total bacterial 16S rRNA genes to provide a relative comparison across the reactors (Fig. 5B). Although methanogens are not bacteria, the mcrA genes quantified were also normalized to bacterial 16S rRNA genes for comparative purposes (Fig. 5C). The ETOH 1 bioreactor contained a notably high percentage of SRB (75%±12%), a result of the total number of bacteria being significantly lower in both ethanol reactors compared to the lignocellulose reactors, whereas the absolute abundance of SRB (not normalized to the 16S rRNA gene) was comparable for all reactors. Thus, the similar sulfate reduction performance of all the bioreactors (Venot et al., 2008) corresponds to the absolute number of SRB present rather than the percentage of SRB in each bioreactor. Methanogens were also significantly lower in the ethanol reactors than in the lignocellulose reactors. Overall, there were no significant differences in total bacteria or the two specific microbial groups among the four lignocellulose-based reactors.
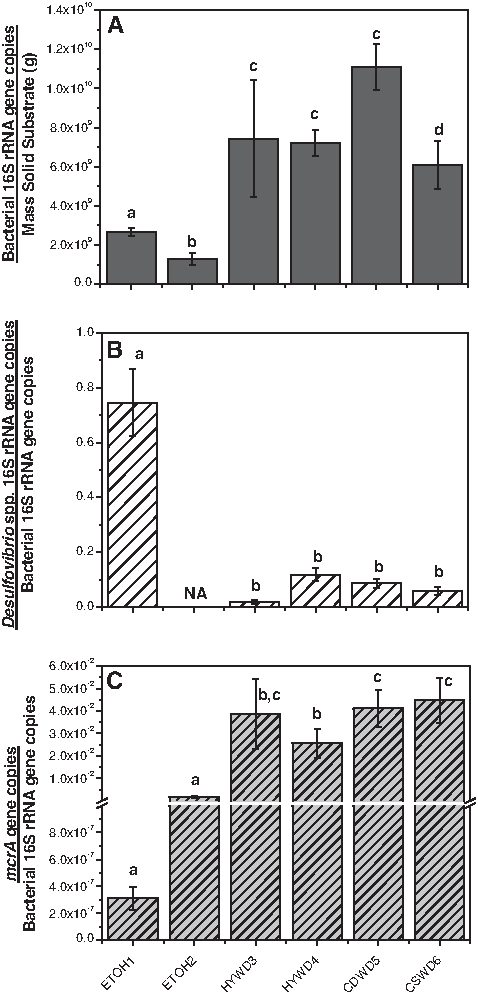
Quantification of total bacteria, sulfate-reducing bacteria of the genus Desulfovibrio, and methanogenic microorganisms for the ETOH, HYWD, and CSWD biochemical reactors by quantitative polymerase chain reaction. Total bacteria 16S rRNA gene copies normalized to mass of substrate
Discussion
Effect of type of substrate on community composition
This study investigated the effect of different organic substrates on the microbial communities in pilot-scale biochemical reactors treating MIW. As expected, it was found that different organic substrates resulted in distinct microbial communities. Key differences were observed between lignocellulose and ethanol-fed reactors in terms of total biomass, overall bacterial diversity, functional groups of bacteria, presence of certain SRB, and quantities of methanogens and SRB. The kinds of microorganisms present, their abundance, and activities have been observed to correspond to critical functional processes and performance within other biochemical reactors (e.g., Pruden et al., 2007; Hiibel et al., 2008; Pereyra et al., 2008), and may dictate reaction rates, ranges of substrates utilized, environmental tolerance range, and resilience to stress.
The overall bacterial diversity was found to be higher in the lignocellulose-based bioreactors (Fig. 1). This was likely a result of the complexity of the lignocellulosic substrate that required a broader array of functional groups for its breakdown compared to ethanol, which could be used directly by the SRB (Tsukamoto and Miller, 1999; Rabus et al., 2000). The differences among the reactors in terms of the kinds of SRB detected were more modest, though there were a few species that were only found in the lignocellulose-based reactors (Fig. 3). Although all SRB conduct dissimilatory sulfate reduction, their substrate utilization capabilities, kinetics, and other characteristics, such as tolerance to oxygen exposure and other stresses, differ (Warren et al., 2005). These factors could have a significant impact on reactor performance. The absolute abundance of the SRB was comparable between the reactors, although the proportion of SRB was higher in the ethanol-based bioreactors due to lower total bacterial biomass (Fig. 5). This was likely impacted by the reactor design in which the abiotic support material (solid limestone and zero valence iron slag) provided a small surface area to mass ratio as compared to the lignocellulosic material, where carbon sources also served as the support material. A few other bacteria besides SRB were detected in the ethanol reactors, including putative fermentative and polysaccharide degrading species (Fig. 2). Their low-level presence is not surprising and is attributed to the horse manure used to inoculate the reactors, which would provide some residual substrate.
Results of both 16S rRNA gene cloning and Q-PCR provided good agreement and indicated that the ethanol-based bioreactors were dominated by SRB (∼70%, Figs. 2 and 5). Ethanol is a simple organic alcohol that can be directly utilized by most species of SRB (Rabus et al., 2000) and was the prevailing carbon source in the ethanol reactors. This created a selective pressure that enriched the microbial community with microorganisms that could utilize this carbon source. By contrast, the low proportions of SRB in the lignocellulose-based bioreactors (∼2%–10%, Figs. 2 and 5) were typical of what has been observed by others (Morales et al., 2005; Hong et al., 2007; Pruden et al., 2007; Pereyra et al., 2008).
Ethanol can also be consumed by some methanogens (Garcia et al., 2000). However, the proportion of methanogens in the ethanol-based bioreactors was lower than in the lignocellulose-based bioreactors (Fig. 5). Several studies have documented the role of electron donor-to-sulfate ratio in governing the outcome of competition between SRB and methanogens (Raskin et al., 1996; Dar et al., 2008). Assuming that methanogens had an overall negative impact on reactor performance due to their competitive relationship with SRB, the low proportion of methanogens might be an advantage of ethanol-based bioreactors. However, methanogens may play a positive role by removing acetate, which is inhibitory to some SRB and under certain conditions (Raskin et al., 1996). Not all SRB are capable of utilizing acetate, in particular D. aerotolerans and D. burkinensis have been reported incapable of doing so when sulfate is the electron acceptor (Mogensen et al., 2005).
Microbial composition and operational upset
During operation, the HYWD 3 and, to some extent, the ETOH 1 bioreactors experienced operational problems that resulted in exposure to the atmosphere. Interestingly, though the exposure occurred several months before sampling, the microbial communities in these reactors presented some corresponding peculiarities. For example, methanogens were found to be lowest in the ETOH 1 bioreactor. Methanogens are strictly anaerobic microorganisms and the exposure to air might have had a detrimental effect on their survival in the reactor, though a similar effect was not observed on methanogen populations in HYWD 3. Rather, detection of Thiobacillus spp. by apsA gene analysis served to indicate the incident of oxygen exposure in the HYWD 3 bioreactors. Thiobacillus spp. are aerobic or denitrifying organisms that convert sulfides to sulfates (Friedrich, 1998), thus reversing the process applied in remediation. The presence of sulfur-oxidizing bacteria is consistent with a study of the field biochemical reactor at the Peerless Jenny King site, which is also periodically exposed to the atmosphere during seasonal low flow (Hiibel et al., 2008). Interestingly, in comparison to the relatively undisturbed CSWD 5 reactor, SRB detected by cloning of 16S genes were comparable in detection frequency (Fig. 2), and only slightly reduced (though not significantly) in terms of Desulfovibrio numbers determined by Q-PCR (Fig. 5) in the HYWD 3 reactor. Instead, cellulose and polysaccharide degraders were noticeably absent from detection by 16S gene cloning. While several cellulose-degrading clostridial species, such as those previously identified in similar columns (Hong et al., 2007), are known to be obligate anaerobes, deterioration of the substrate during the temporary aerobic conditions may be an additional factor negatively impacting these groups.
Implications for response to environmental stress
Interestingly, the performance of the six biochemical reactors was comparable over the time-scale of the present study, though the ethanol reactors offered some advantage in terms of overall sulfate and metals removal (Venot et al., 2008). The results of this study indicate that different microbial compositions and diversities can produce consistent sulfate and metal removal. This, however, raises the question of how these communities would perform in response to repeated environmental stressors, beyond the cold winter and reactor upset observed in the one season in this study. In full-scale situations, reactors are repeatedly exposed to below-freezing temperatures during winter, overloads of sulfate and metals during spring runoff, and exposure to air, among others. These factors are known to have an effect on the microorganisms and, thus, on the remediation performance, as recently reported in the case of toxic loads of heavy metals (Gibert et al., 2008). The higher microbial diversity of the lignocellulose-based bioreactors might represent an important advantage in terms of resilience to stress. Several studies have cited the importance of diversity in community stability. For example, Koschorreck et al. (2010) reported that increased microbial diversity in a microbial biofilm treating acidic mine pit lake water is an important factor in stabilizing the biofilm's function under fluctuating and partly oxidizing conditions in the reactor. The increased diversity is thought to provide a reservoir of strains with complementary ecological niches (von Canstein et al., 2002). Interestingly, the HYWD 3 bioreactor was much lower in overall diversity than the CSWD 5 bioreactor that was not exposed to oxygen (Fig. 1), indicating that stress events themselves can lead to a decrease in diversity. Given the regularity of changing environmental conditions in reactors and the potential effects that they can have on performance, the temporal relationship between microbial community and stress in biochemical reactors should be further explored to provide insight for future installments.
Summary
This study provides important insight about the effects of organic carbon substrate on the microbial community present in sulfate-reducing bioreactors treating MIW. These microbial communities are of special interest because they are the ultimate catalyst of biological sulfate reduction driving remediation of the MIW. The following are the key findings of this study:
• Profiling of 16S rRNA and apsA genes, along with Q-PCR of Desulfovibrio-Desulfomicrobium provided highly complementary and congruent information about the microbial communities. • Ethanol-based substrates result in strong enrichment of SRB (∼70%) relative to lignocellulosic-based bioreactors (∼2%–3%). • Broad similarities were observed among the reactors in terms of the numbers (only three to four species) and kinds of SRB observed, though there were a few SRB that were only detected in lignocellulosic-based bioreactors. • Overall bacterial diversity was higher in lignocellulosic-based bioreactors. • Methanogenic populations were significantly lower in ethanol-based bioreactors. • Upset of a lignocellulosic-based bioreactor in terms of exposure to oxygen corresponded with an enrichment of sulfur oxidizing bacteria, an undesirable microbial group that reverses the desired sulfate reduction-based remedial process. Upset also appeared to be more detrimental to anaerobic cellulose-degrading bacteria than to SRB, which are traditionally thought to be the population of concern after oxygen exposure.
The above-noted attributes may have important implications on the choice of organic substrate for these types of biochemical reactors. Assuming that reactor upset is inevitable, given the remote location of field sites and lack of resources for active monitoring and maintenance, which is better—ethanol or lignocellulose? The overall paucity in microbial diversity in the ETOH bioreactors suggests that they may be disadvantageous in this regard and could experience a dramatic crash in the microbial community in response to repeated stress. However, the case could also be made that ethanol-based biochemical reactors will perform better in the long-term because the diversity of the SRB, which are most critical to reactor function, was about the same in both kinds of reactors. The SRB actually appeared to recover better from stress relative to cellulose degrading bacteria, thus excluding upstream groups involved in lignocellulose degradation through addition of ethanol may avoid the complicating factor of a more complex upstream microbial community. Also, undesirable Thiobacillus spp. appeared to establish themselves better in the lignocellulosic reactor (HYWD 3) than in the ethanol biochemical reactor (ETOH 1) that experienced upset. A study aimed at determining the effect of microbial community and SRB diversity on reactor performance in the face of repeated aerobic stress would be of value in extending the observations of this study.
Footnotes
Acknowledgments
This work was supported by the EPA Office of Research and Development's Engineering Technical Support Center, under contract with Golder Associates, Inc., Contract # GS-10F00130N DO #1101 (David Reisman, Project Officer; reisman.david@epa.gov), and by the Water Research Experience for Undergraduates program at CSU sponsored by the National Science Foundation. Funding was also provided by the National Science Foundation Award CBET-0651947; the findings do not necessarily represent the opinions of NSF. This research has undergone EPA peer and policy review, and has been cleared for publication. Mention of commercial products does not constitute endorsement by the Agency.
Author Disclosure Statement
The authors do not have any commercial or professional conflicts of interest associated with the contents of this article.