
Abstract
Abstract
We examined the potential formation of chloropyromorphite [Pb5(PO4)3Cl(s)] as an experimental artifact in the physiologically based extraction test (PBET), a common surrogate method used to assess the effectiveness of in situ phosphate-based treatment to reduce Pb bioavailability in soil. Although the conditions of the PBET are strongly thermodynamically favorable for the formation of Pb5(PO4)3Cl(s) due to the high concentrations of chloride (0.25 M) in the extraction solution, Pb precipitation was minimal (<5%) over the normal 1-h duration of the PBET. Lead concentrations in the PBET fluid did, however, decrease by 70%–85% over 5–7 days. Precipitation rates increased sharply as the glycine concentration was decreased from 0.4 M (standard PBET) to 0 M, approaching the rapid Pb5(PO4)3Cl(s) precipitation rates typically observed in aqueous solutions. Thus, we conclude that glycine substantially lowers the precipitation rate of Pb5(PO4)3Cl(s), and at the standard 0.4 M glycine concentration of the PBET, insignificant formation of Pb5(PO4)3Cl(s) will occur in the extraction fluid itself (i.e., there will be no experimental artifact) over the normal duration of the test. Further, the concentration of Pb stabilized in our solutions at an apparent solubility significantly exceeding what would be predicted thermodynamically (i.e., log Kso apparent of −79.5±0.9 vs. a reported value of −84.4). In sum, we conclude that significant formation of Pb5(PO4)3Cl(s) does not occur in the PBET, supporting its use in assessing the effectiveness of in situ phosphate immobilization. Our results also support the emerging body of evidence that the thermodynamic potential for Pb precipitation via the formation of Pb5(PO4)3Cl(s) may not be as great as previously believed.
Introduction
Lead, phosphate, and chloride (Cl−) readily react in solution to form Pb5(PO4)3Cl(s) (Zhang et al., 1997; Zhang and Ryan, 1998, 1999a, 1999b; Manecki et al., 2000; Scheckel and Ryan, 2002; Odyakov and Zhizhina, 2005). In many cases, however, direct evidence for the formation of Pb5(PO4)3Cl(s) in P-amended soil is lacking (Scheckel et al., 2005). A common (Berti and Cunningham, 1997; Hettiarachchi et al., 2000, 2001, 2003; Tang et al., 2004; Sonmez and Pierzynski, 2005; Yoon et al., 2007) though indirect method of assessing the effectiveness of P-based in situ remediation is the physiologically based extraction test (PBET), an extraction test designed to measure the amount of soluble and, thus, potentially bioavailable Pb in a child's gastrointestinal tract (Ruby et al., 1996). The PBET, an in vitro extraction protocol that has been widely used to estimate changes in Pb bioaccessibility induced by soil amendments, was used as a surrogate for oral bioavailability.
However, Scheckel et al. (2003, 2005) recently warned that rather than forming in situ, Pb5(PO4)3Cl(s) may actually be forming during the extraction test itself (i.e., an experimental artifact), which has caused some researchers (Hettiarachchi et al., 2000, 2001; Ryan et al., 2001) to interpret their own results with caution. Although perhaps not recognized by some researchers, such an artifact would be of particular concern with the PBET due to the very high concentration of Cl (0.25 M) in the extraction solution, which greatly increases the thermodynamic potential for the formation of Pb5(PO4)3Cl(s). To the extent that the reported reductions in bioaccessibility, a surrogate for oral bioavailability, are a result of an experimental artifact rather than bona fide in situ treatment, the efficacy of P-based amendments in reducing Pb bioavailability becomes uncertain.
The primary objective of this study was to evaluate the potential formation of Pb5(PO4)3Cl(s) as an experimental artifact during the PBET. To accomplish this objective, we monitored Pb precipitation and the saturation indices of Pb5(PO4)3Cl(s) and other minerals in the PBET and various control solutions as a function of time over a period of 1 week. These results are important both in interpreting existing data and in determining the future use of the PBET in assessing the effectiveness of P-based remediation.
Materials and Methods
All chemicals employed in this study were analytical grade or better and obtained from Fisher Scientific (Pittsburgh, PA), and all solutions were prepared with deionized water (18 MΩ·cm) from an ion exchange apparatus. The extraction test that we examined was a streamlined version (Kelley et al., 2002) of the original PBET method (Ruby et al., 1996), which was designed to replicate the solubility-limiting conditions of a child's gastrointestinal tract. Although the original PBET included both simulated stomach and small intestine extraction phases, recent research has suggested that Pb dissolution in the simulated stomach phase alone is predictive of Pb bioavailability in animals (Kelley et al., 2002). Similarly, although glycine was not used in the original PBET, the streamlined version was modified to include glycine in lieu of other organic ligands (Kelley et al., 2002). In particular, the primary factor controlling Pb bioaccessibility is the simulated stomach pH (Ruby et al., 1996; Oomen et al., 2002; Yang et al., 2003; Ryan et al., 2004). The PBET was originally designed to simulate a fasting child's stomach at a pH of 1.5. However, Ruby et al. (1999) noted that both a pH of 1.3 and 2.5 in the stomach phase of the in vitro test correlated equally well with the relative bioavailability of the in vivo weanling rat model (pH 1.5 was ultimately selected). Another recent Pb bioavailability study has indicated that an in vitro pH of 2.3 correlates well (r2=0.90) with in vivo rat bone results (Brown et al., 2003). The results of other recent research further indicate that an extraction pH of ∼2.3 more accurately reflects reductions in Pb bioavailability due to P amendments (Brown et al., 2003, 2004; Ryan et al., 2004). We, thus, used an extraction pH of 2.3 to reflect the results of this recent research.
The PBET is an extractor submerged in a water bath heated to body temperature (37°C±2°C) and connected to an external motor that rotates the samples end-over-end at 30±2 rpm. A 0.1 g soil sample, 1:100 solid and solution ratio, which has previously been shown to be consistent with a 1.0 g sample at the same solid-to-solution ratio (Yang et al., 2002), was used for running the PBET. A detailed description of our PBET system has been reported in Yang et al. (2002). The PBET is nominally a 1-h extraction, although we also collected samples over both shorter and longer periods of time as well. In addition to the standard PBET chemical extraction solution (described below), 0.241 mM Pb(NO3)2 and 16.1 mM KH2PO4 were added to the solution. In terms of total Pb and P, these concentrations are equivalent to adding 1 g of soil with 5,000 mg kg−1 Pb amended with 5% P to 100 mL of PBET solution as specified in the test. The samples were removed at desired intervals, centrifuged, and filtered with a 0.45 μm filter. Lead concentrations in the PBET solution were measured with a flame atomic absorption spectrometer. Thermodynamic calculations were performed with Visual Minteq (v. 2.3.2). The model was run at a fixed pH of 2.3 and an ionic strength of 0.25 M. The Pb(NO3)2, KH2PO4, and NH2CH2COOH (Hglycine) concentrations were added in the model in terms of Pb2+, NO−3, K+, H+,
In addition to experiments at body temperature, we also conducted extractions at room temperature (∼22°C) so that the results would be more directly comparable to thermodynamic calculations, where the majority of the data is available at 25°C. The PBET solution is a 0.4 M glycine solution adjusted to the desired pH with concentrated HCl. Since glycine is a buffer, high concentrations of HCl (0.25 M for 0.4 M glycine) are required to adjust the solution to pH 2.3, and the resulting high concentrations of Cl greatly increase the thermodynamic potential for the formation of Pb5(PO4)3Cl(s) in the extraction solution. To examine the role of both Cl and glycine in reactions of Pb in the PBET solution, we also conducted some experiments with HNO3 rather than HCl and with varying concentrations of glycine. In all experiments, NaCl and HNO3 were added as necessary to maintain the same pH, Cl concentration, and ionic strength of the original PBET solution. The composition of each of the solutions is given in Table 1.
PBET, physiologically based extraction test.
Results and Discussion
During the standard PBET (1 h, 37°C, pH 2.3, 0.4 M glycine), we observed minimal (<5%) Pb precipitation from solution (not shown), a surprising result given the reported rapid precipitation kinetics of Pb5(PO4)3Cl(s) (Nriagu, 1974; Scheckel and Ryan, 2002) and the tremendous thermodynamic driving force for the reaction for the formation of Pb5(PO4)3Cl(s). The initial saturation index (SI) for the formation of Pb5(PO4)3Cl(s) via
was 6.2. The SI is the logarithmic (base 10) ratio between ion activity product (Q) and the solubility constant (Kso). For equation (1), the SI is written as follows:
A SI of <0 indicates that the system is undersaturated with regard to the solid. A SI of 0 indicates that the solution is in equilibrium. An SI of greater than 0 indicates that the system is supersaturated with regard to the solid.
To ascertain the reason for the lack of Pb precipitation observed during the standard PBET, we conducted additional experiments over a longer time period and using both HCl (standard PBET) and HNO3 to adjust the pH to 2.3. These supplemental experiments were also conducted at room temperature (∼22°C) so that the results could be modeled thermodynamically, as most of the thermodynamic data are available at 25°C. The concentrations of Pb over time in both the HCl-based and HNO3-based solutions are shown in Fig. 1a and b. Over the first hour (the normal length of the PBET), the concentrations of Pb were reduced by only ∼5% and ∼10% in the HCl-based and HNO3-based PBET solutions, respectively. This relatively small decrease in solution-phase Pb over the normal 1-h duration of the PBET indicates a limited potential for the formation of solid-phase Pb that could be misconstrued as an in situ reduction in Pb bioaccessibility (e.g., an experimental artifact). After decreasing by ∼10% over the first hour, the Pb concentration in the HNO3-based PBET did not significantly (p>0.05) decrease further over the remaining 7 days of the experiment. In contrast, the Pb concentration in the HCl-based PBET solution continued to decrease for 5 days before reaching steady-state. Although the SI of Pb5(PO4)3Cl(s) in the HCl-based solution decreased over the course of the experiment, even after 7 days (Fig. 1b), when the Pb concentration had stabilized, the SI was still 4.4. The difference in the Pb concentration profiles between the two PBET solutions illustrates that Cl is involved in the precipitation process (there was no Cl in the HNO3-based solution), consistent with the long-term formation of Pb5(PO4)3Cl(s) in the HCl-based PBET solution.
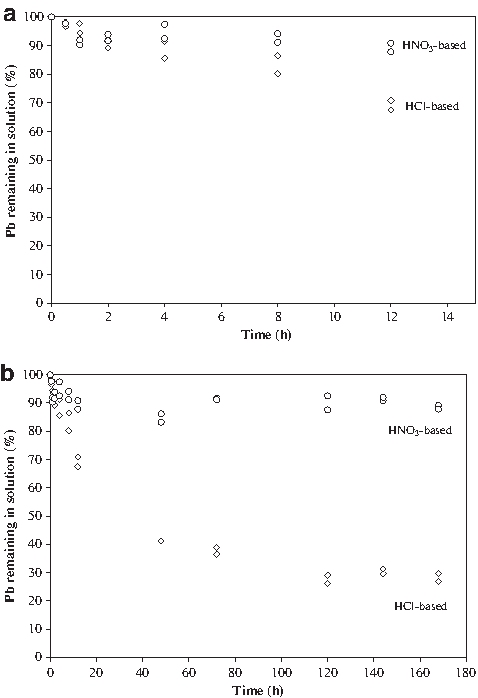
Using a HNO3-based rather than the normal HCl-based PBET solution eliminated both the potential for the formation of Pb5(PO4)3Cl(s) and most of the actual Pb precipitation. Over the course of the experiment, both solutions remained undersaturated with regard to PbHPO4(s) (−1.5<SI <−0.5) and well undersaturated (SI <−2) with regard to other Pb-containing solids. The small (∼10%) but significant (p<0.05) Pb precipitation observed in the HNO3-based PBET solution may have been due to the precipitation of PbHPO4(s), although the solution remained slightly undersaturated (SI∼−0.6) with regard to PbHPO4(s) over the course of the experiment. Ionic strength effects (I=0.26 M), potential differences in solubility due to varying crystallinity, particle size, and so on, and uncertainties in Kso obviate overly rigid interpretation of thermodynamic calculations. Sauve et al. (1998), for example, noted that Pb5(PO4)3Cl(s) (and other solid phosphates as well) solubility studies are few and that the existing studies have largely been conducted under a limited range of conditions.
Although significant Pb precipitation from the HCl-based extraction solution was observed over a 5–7-day period, these results are not consistent with other reports of very rapid (seconds to minutes) precipitation of Pb5(PO4)3Cl(s) from aqueous solutions (Nriagu, 1974; Scheckel and Ryan, 2002). Accordingly, we examined the effect of glycine on the rate of precipitation of Pb5(PO4)3Cl(s) from solution. As the glycine concentration decreased from 0.4 M (standard PBET) to 0 M, the Pb precipitation rate increased sharply (Fig. 2a, b). As just noted, with 0.4 M glycine, only ∼5% of the Pb was removed from the extraction solution after the normal PBET duration of 1-h (Fig. 2a) and Pb concentration continued to decrease until reaching a steady-state level after ∼5 days (Fig. 2b). In contrast, in the absence of glycine, a white precipitate (later identified as Pb5(PO4)3Cl(s) by X-ray diffraction) was observed immediately, and ∼50% of the Pb was removed from solution before the first sample could be taken (<1 min) with no additional Pb precipitation observed over the next 7 days. In the presence of just 0.001 M glycine, the Pb concentration reached a steady state after ∼8 h, remaining approximately constant over the remainder of the 7-day experiment. We were not able to definitively identify any Pb5(PO4)3Cl(s) in the X-ray diffraction patterns of the solids precipitated from the glycine-containing solutions. Thus, we conclude that the presence of glycine kinetically restricts the formation of Pb5(PO4)3Cl(s) from the extraction solution.
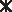
), 0.001 (□), 0.01 (+), 0.2 (X), 0.3 (Δ), and 0.4 M (⋄)] and time (0–30 h; T=∼22°C). ), 0.001 (□), 0.01 (+), 0.2 (X), 0.3 (Δ), and 0.4 M (⋄)] and time (0–180 h; T=∼22°C).
This kinetic effect could be due, at least partially, to glycine complexation with Pb. Thermodynamically, 55% of the aqueous Pb was bound in complexes with glycine in the 0.4 M glycine solution compared with <1% of the Pb bound in glycine complexes in the 0.001 M glycine solution (Fig. 3). The major Pb-chloride complexes include PbCl+, PbCl2(aq), PbCl3−, and PbCl4−. The major Pb-glycine complexes include PbH-Gly2+, and PbH2-(Gly)22+. The percentage of glycine complexation with Pb was calculated from the known stability constants of Pb-glycine complexes using Visual Minteq (Table 2a) and not determined directly by measurement. However, these results are consistent with those of other studies (Lang and Kaupenjohann, 2003; Scheckel and Ryan, 2003; Hashimoto et al., 2009) that reported the presence of natural organic ligands, particularly at low pH, which kinetically inhibited the formation of Pb5(PO4)3Cl(s) by impairing crystallization and inhibiting crystal growth. Similarly, natural organic matter has also been found to impair the development of other lead containing solids during the corrosion of lead metals used in plumbing studies (Korshin et al., 2005).
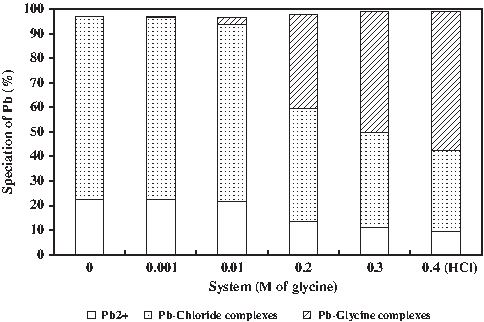
Speciation of dissolved Pb in PBET systems with differing glycine concentration. Results were calculated using Visual Minteq.
From NIST Standard Reference Database 46 (www.nist.gov/srd/nist46.cfm).
Over the 7-day duration of the experiments, Pb concentrations stabilized after ∼50%–85% precipitation. However, thermodynamically, >95% precipitation should have been observed in all cases due to the precipitation of Pb5(PO4)3Cl(s). With the exception of Pb5(PO4)3Cl(s), the solutions were well undersaturated (SI<−0.8) with regard to all other solids phases at the end of the experiment. The SI and apparent solubility products of Pb5(PO4)3Cl(s) at the end of the experiments in the HCl-based solutions are shown in Table 2b. The SI for Pb5(PO4)3Cl(s) at the end of the experiment was >3.9 for all solutions, indicating that the solutions were still quite supersaturated with regard to Pb5(PO4)3Cl(s). The observed apparent log Kso ranged from −78.1 to −80.5 with an average of −79.5±0.9. The possible minerals considered for saturation are listed in Table 3 along with their log Kso values.
Other recent papers have also reported Pb remaining in solution well in excess of what would be predicted thermodynamically from the precipitation of Pb5(PO4)3Cl(s) (Stanforth and Qiu, 2001; Yang et al., 2001). Zhang and Ryan (1999b) studied the formation of Pb5(PO4)3Cl(s) from cerrusite [PbCO3(s)] and hydroxyapatite [Ca5(PO4)3OH(s)]. However, even in systems where there was reportedly complete transformation of PbCO3(s) to Pb5(PO4)3Cl(s), the resulting equilibrium Pb concentration was still well above (at least an order of magnitude in some instances) what would be expected thermodynamically, consistent with the recent analysis of their data by Martínez et al. (2004). Our own analysis of Zhang and Ryan's (1999b) data (not shown) indicates that the reported equilibrium Pb concentrations are markedly more consistent with a log Kso of approximately −79.1 than of a log Kso of −84.4. Similarly, Xie and Giammar (2007) recently reported that their pH-dependent solubility data were more consistent with a Pb5(PO4)3Cl(s) log Kso of −80.4 vice −84.4. Although Xie and Giammar (2007) noted that the reported log Kso of −84.4 was measured under rather limited and atypical conditions (pH ∼2.3 and 0.1 M NaCl), those conditions are remarkably similar to the ones of our experiments.
Summary and Conclusions
The in situ remediation of Pb-contaminated soils via P-based amendments has been extensively studied and sometimes championed over the last 15+ years, and the PBET has seen considerable use in measuring the resulting decreases in Pb bioaccessibility. However, despite the reported rapid precipitation kinetics of Pb5(PO4)3Cl(s) from aqueous solution and the strong thermodynamic driving force in the PBET solution, we observed negligible Pb precipitation from solution under the normal conditions of the PBET (1 h, 37°C and 0.4 M glycine). Thus, the reported reductions in Pb bioaccessibility measured by the PBET (and potentially PBET-like extractions) are unlikely to be the result of experimental artifacts. Thermodynamic limitations, the high dosages of phosphate required to achieve sometimes modest reductions in Pb bioaccessibility (Moseley et al., 2008), attendant concerns over soil structure and eutrophication (Scheckel and Ryan, 2004; Yoon et al., 2007), and mobilization of cocontaminants and even background arsenic (Kilgour et al., 2008) may warrant reexamination of the utility of P-based amendments as in situ remediation technology.
Footnotes
Acknowledgment
This research was sponsored by the Strategic Environmental Research and Development Program under the direction of Dr. Andrea Leeson.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.