
Abstract
Abstract
Sorption kinetics and controlling factors of phenanthrene (PHE) adsorption on limestone were obtained by batch experiments. A pseudo-first-order equation fit well with the experimental data. Roles of carbonate and organic matter in the adsorption process of PHE were studied by using a sequential separation procedure. It was discovered that organic matter remarkably affected the behavior of PHE in the adsorption procedure, and carbonate slightly affected the adsorption process due to its influence on physical characteristics of limestone. Different pH conditions led to precipitation and dissolution of carbonate and dissociation of limestone hydroxyl groups, which affected adsorption capacity. In the presence of low ionic strength, calcium ions would occupy some adsorption sites, leading to a decrease in PHE adsorption. By contrast, standard free energy (ΔG0) became more negative when the concentration of CaCl2 increased from 0.005 to 0.1 M, which was consistent with the amount of adsorption increase. The influence of humic acids (HA) was also investigated. There was competitive adsorption between PHE and HA. The affinity of HA for limestone was stronger than that of PHE, which when adsorbed on limestone could be desorbed by HA added later. Finally, the coexistence of PHE and acenaphthylene indicated that there was a competitive adsorption between them, although it was easier for PHE to be adsorbed on limestone.
Introduction
P
Karst massifs contain large amounts of high-quality groundwater resources for drinking water supply of the human society and more than 25% of the world's population lives in karst areas (Kovačič and Ravbar, 2013). With their relatively high surface connectivity and groundwater flow rates, karst aquifers are particularly vulnerable to be contaminated from surface water (Simmleit and Herrmann, 1987). Sorption/desorption of hydrophobic organic contaminants such as PAHs play a major role in controlling their fate and transport in the aquatic environment (Low and Batley, 1988) and participate in mineral precipitation and dissolution (Findlay et al., 2003). Although there have been a lot of reports about sorption of heavy metals on limestone (Aziz et al., 2008; Sdiri et al., 2012), little work has been done to characterize the interaction between PAHs and carbonate minerals that are major mineral phases in karst aquifers. Therefore, studying PAHs sorption on limestone can improve our understanding of the geochemical behavior of PAHs in karst hydrologic systems.
According to the results of our geochemical survey in 2013 in the Guozhuang karst water system of Shanxi province in northern China, the concentration range of phenanthrene (PHE) was 742–3740 ng/L in surface water, 1039–5069 ng/g in topsoil, and 400–1306 ng/L in karst groundwater, all of which were higher than those published elsewhere (Helaleh et al., 2005; Xu and Lee, 2008; Han et al., 2013). Surface water leakage and topsoil leaching are the major causes for groundwater contamination at Guozhuang (Han et al., 1993). For the transport of PHE in the karst aquifers, its sorption on limestone may have significance impact.
However, there have been few studies that specifically examine PHE-carbonate kinetics in karst water systems. The objectives of our study are as follows: (1) to characterize the sorption kinetics of PHE on limestone; (2) to determine the linear absorption coefficient (Kd) of PHE at the limestone–water interface; (3) to understand the influence of solution chemistry, humic acids (HA), and coexisting PAHs on PHE sorption.
Materials and Methods
Adsorbents
Limestone samples were collected from the karst area of Guozhuang karst water system in Shanxi province, China (Fig. 1). The total PAHs concentration in places from which limestone samples were collected ranged from 206.7–1666.7 ng/L in groundwater, and 98.9–3966.3 ng/g in topsoil. Samples were sealed in plastic bags and stored at −4°C before use. For sorption experiments, total organic carbon (TOC), and X-ray diffraction (XRD) analysis, the samples were ground and passed respectively through 100 and 200 mesh sieve.

Limestone sampling sites at Guozhuang karst water system.
Chemicals
PHE and acenaphthylene (ACE) were purchased from AccuStandara, and prepared in methanol at varying concentrations. Other organic solvents included methanol, dichloromethane (DCM) (analytical grade; CNWBOND). HCl, NaOH, sodium humate, CaCl2, and NaN3 were purchased from Sigma-Aldrich. Anhydrous sodium sulfate was heated at 450°C in a muffle furnace for 4 h and stored in a sealed desiccator prior to use. All glassware were cleaned with sulfuric acid potassium dichromate solution, washed with water, and dried at 180°C for 4 h.
Separation procedures
Separation procedures were performed sequentially to get S1 and S2 fractions from limestone (Yang et al., 2011).
1. S1 fractions were obtained by removing carbonate from original limestone (S0) using HCl repeatedly until there was no production of carbon dioxide. Then, the sample was thoroughly washed with deionized water until the solution pH value was neutral.
2. After removing carbonate, 30% H2O2 solution was added dropwise to S1 fractions to remove the organic carbon (OC), and the mixture was intermittently agitated at 40°C and then evaporated. This step was repeated three times in 48 h to obtain S2 fractions. All samples of different fractions were air dried and ready for sorption experiments.
Sorption experiments
Limestone (0.5 g) was mixed with 20 mL of varying concentrations of PHE (50–500 μg/L) solution, which contained 0–0.2 M CaCl2 to adjust ionic strength and 200 mg/L NaN3 to restrain microbial activities. Controls without adsorbent were determined in the same way to account for PHE loss by the glass wall and cover. The methanol concentration of PHE aqueous solutions was less than 0.1% (v/v) to prevent cosolvent-effect (Qian et al., 2011). Then, the centrifuge glass tubes were packed with tinfoil to prevent photodegradation and shaken in a shaking waterbath (HY-5; Guohua Company) at 120 rpm (20°C) for 12 h to achieve sorption equilibrium. After sorption equilibrium, aqueous solution and solid phase were separated through a centrifuge (TG16-WS; Xiangzhi Company) at 4000 rpm for 5 min. Then, 15 mL supernatant was extracted in 10 mL DCM by shaking for 5 min for three times. Organic phase was collected in a 50 mL flask. The final volume was 0.2 mL after rotary evaporation and nitrogen flush.
Analytical method
TOC of the sorbents (S0, S1 and S2) was determined by LiquiTOC analyzer (Elementar) after treating with dilute HCl acid to remove carbonates. Powdered limestone samples were examined at room temperature using a Philips X'Pert Pro X-ray diffractometer with a Cu tube, graphite crystal monochromator and proportional counter (X'Pert ProDY2198; PANalytical). The specific surface area and pore-size distribution were measured by nitrogen adsorption using the Brunauer-Emmett-Teller (BET) method with an ASAP 2020M surface area analyzer (Micrometrics Instrument Corporation). Fourier transform infrared (FTIR) spectra of the sorbent S0 under different pH conditions as KBr pellets were recorded with a Thermo Fisher Nicolet 6700 FTIR spectrometer. The spectra were collected in transmission mode at a resolution of 4 cm−1, and 100 scans were recorded in the 4000–400 cm−1 range to ensure good signal to noise ratio.
Analysis of PHE and ACE concentration was performed on a high-performance liquid chromatography (HPLC) system (HP1100; Aglient) with a HPLC pump, a 474 scanning fluorescence detector, and a 2347 double beam UV detector. The separating column was a ChromSep C18 column (250×4.6 mm, particle size 5 μm; Varian), the HPLC separation was carried out with 90% methanol and 10% water as mobile phase at a flow rate of 1 mL/min at 30°C, the monitoring wavelength was 254 nm. HA was measured by UV/visible spectrophotometer (Hitachi U-3900) at 254 nm.
The amount of PHE adsorbed by adsorbent at time t (qt, μg/g), and the amount of PHE adsorbed by adsorbent at equilibrium (qe, μg/g), were calculated by the two equations given below:
where C0 and Ce (μg/L) are the initial and the equilibrium concentrations of PHE in solutions, Ct (μg/L) is the concentration of PHE at time t, V (L) is the volume of the solution, and m (g) is the mass of adsorbent used.
Sorption kinetics is critical in governing the solute uptake rate and sorption efficiency of an adsorbent. Therefore, it is important to predict the rate at which pollutant is removed from aqueous solution to design appropriate sorption treatment schemes. The typical kinetics models normally consider both the external and internal mass transfers. Examples of these models are film-pore diffusion, film-surface diffusion, pore diffusion, surface diffusion, and combined pore and surface diffusion models. Further, mass transfers of sorption often involve different mechanisms. Therefore, for the simplicity and practical use of engineering applications, the global kinetics expressions such as the Lagergren pseudo-first-order and pseudo-second-order equations, have been commonly used.
The pseudo-first-order rate expression of Lagergren may be written as (Lagergren, 1898):
where qe and qt are the amounts of PHE adsorbed on limestone at equilibrium and time t, respectively (μg/g), and k1 is the rate constant arising from the first-order model (h−1).
The pseudo-second-order kinetics model may be expressed by the following (Ho and McKay, 1999):
where k2 (g/μg per hour) is the rate constant of the second-order sorption.
Results and Discussion
Characterization of sorbents
XRD results showed that the parent limestone sample mainly consist of calcite (around 90%), and a small amount of dolomite, gypsum, and quartz. S1 and S2 mainly consist of the amorphous materials, containing very small amounts of calcite, dolomite, and quartz (Fig. 2). These results suggest that all calcium carbonate was removed by HCl. The BET surface area of S0, S1, and S2 is 35.3, 40.9, and 38.2 m2/g, respectively. Thus, the sorbents have desirable surface areas. The average pore size and total pore volume of S0, S1, and S2 was observed to be 66.9, 65.2, 66.3 Å, and 0.58, 0.66, 0.63 cm3/g, respectively. The maximum dimension of a PHE molecule in aqueous solution is 10 Å (Huang et al., 1996). This may facilitate increased dispersion of PHE in the inner layer of the granular limestone. The FTIR spectrum of limestone (S0) is shown in Fig. 3. Limestone sample was dominated by the large band at 3440 and 1050 cm−1, which represented the stretching vibration of hydroxyl groups and CH2 units, respectively. Peak at 1700 cm−1 was assigned to the formation of carboxy groups, and band at 796, 693, 535, and 470 cm−1 were considered to be crystalline phase vibration absorption peaks.
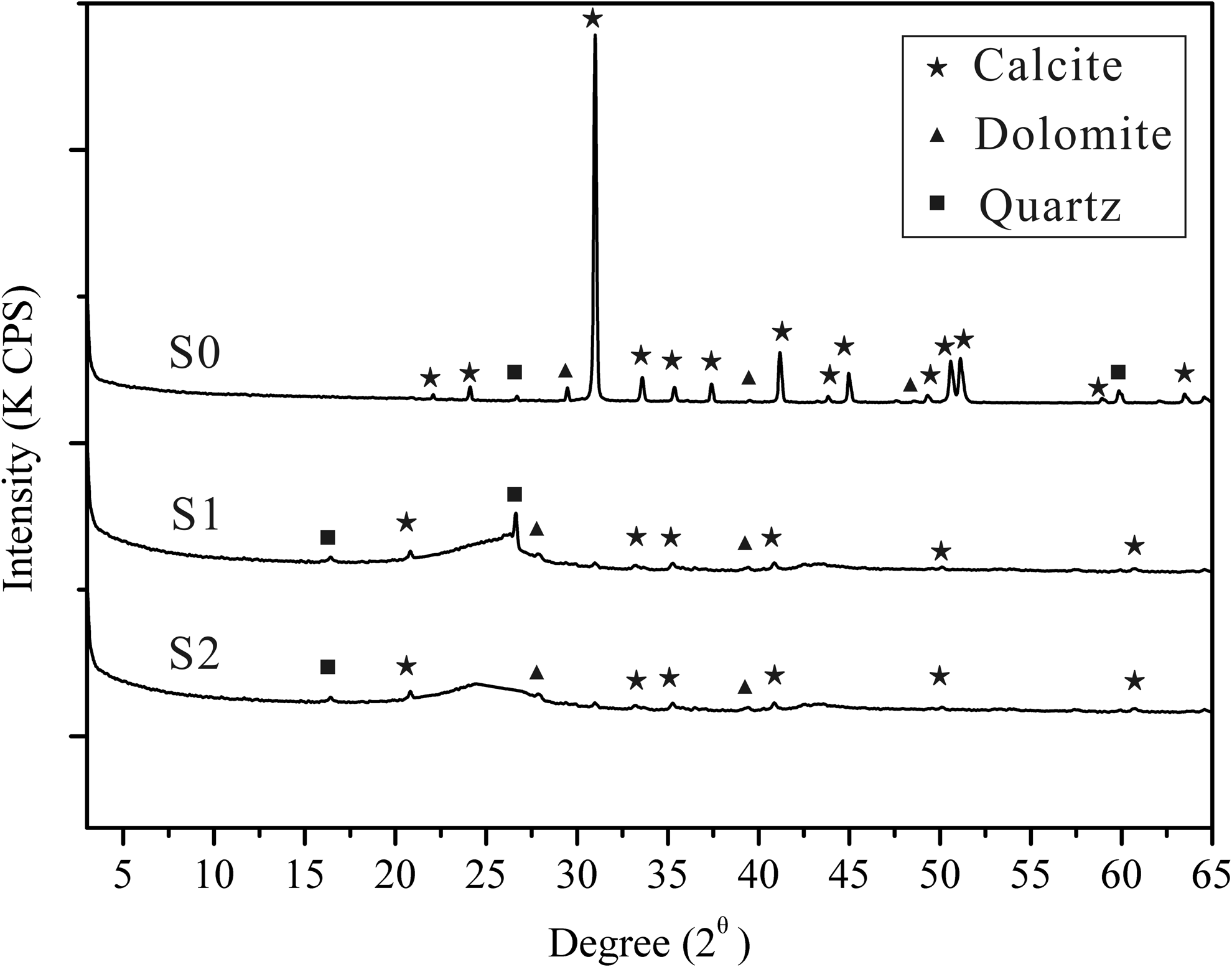
X-ray diffraction pattern of the limestone sample (S0, S1, and S2).
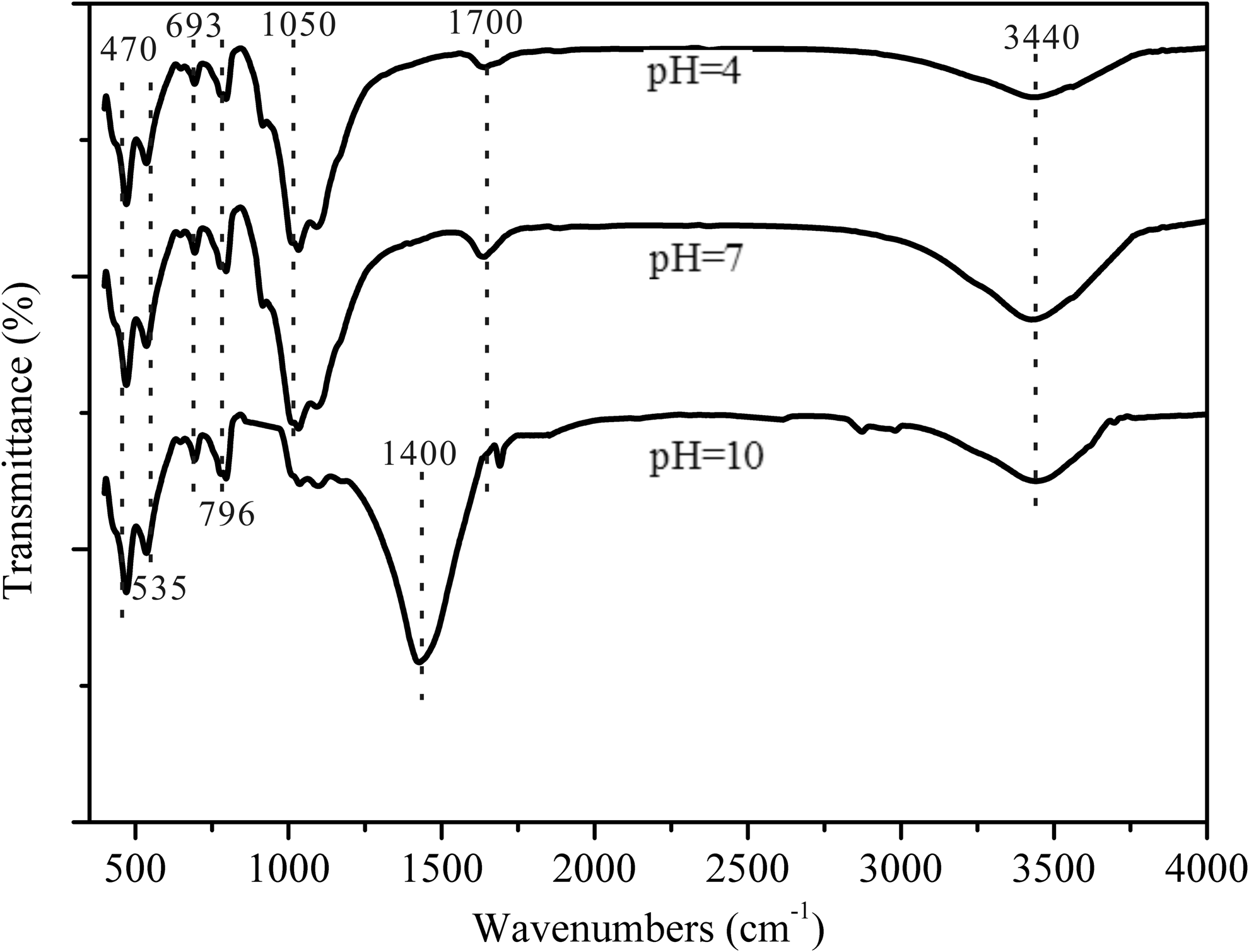
Fourier transform infrared spectra of limestone sample under different pH.
Sorption kinetics of PHE on limestone
Rate constants and equilibrium sorption capacities of two kinetics equations were calculated by means of linear curve fitting analysis. A comparison of results with the correlation coefficients (R2) is listed in Table 1. Comparing values of R2, qt and qe for pseudo-first-order and pseudo-second-order equations, the former was better than the latter (Fig. 4). Therefore, the pseudo-first-order kinetic model was suitable to describe the kinetics adsorption process of PHE on limestone, which seemed to suggest a one-site-occupancy adsorption when the adsorbing molecule reacts with one adsorption site (Ho et al., 2000). Particle diffusion was the mechanism controlling the rate of the PHE adsorption process on limestone.
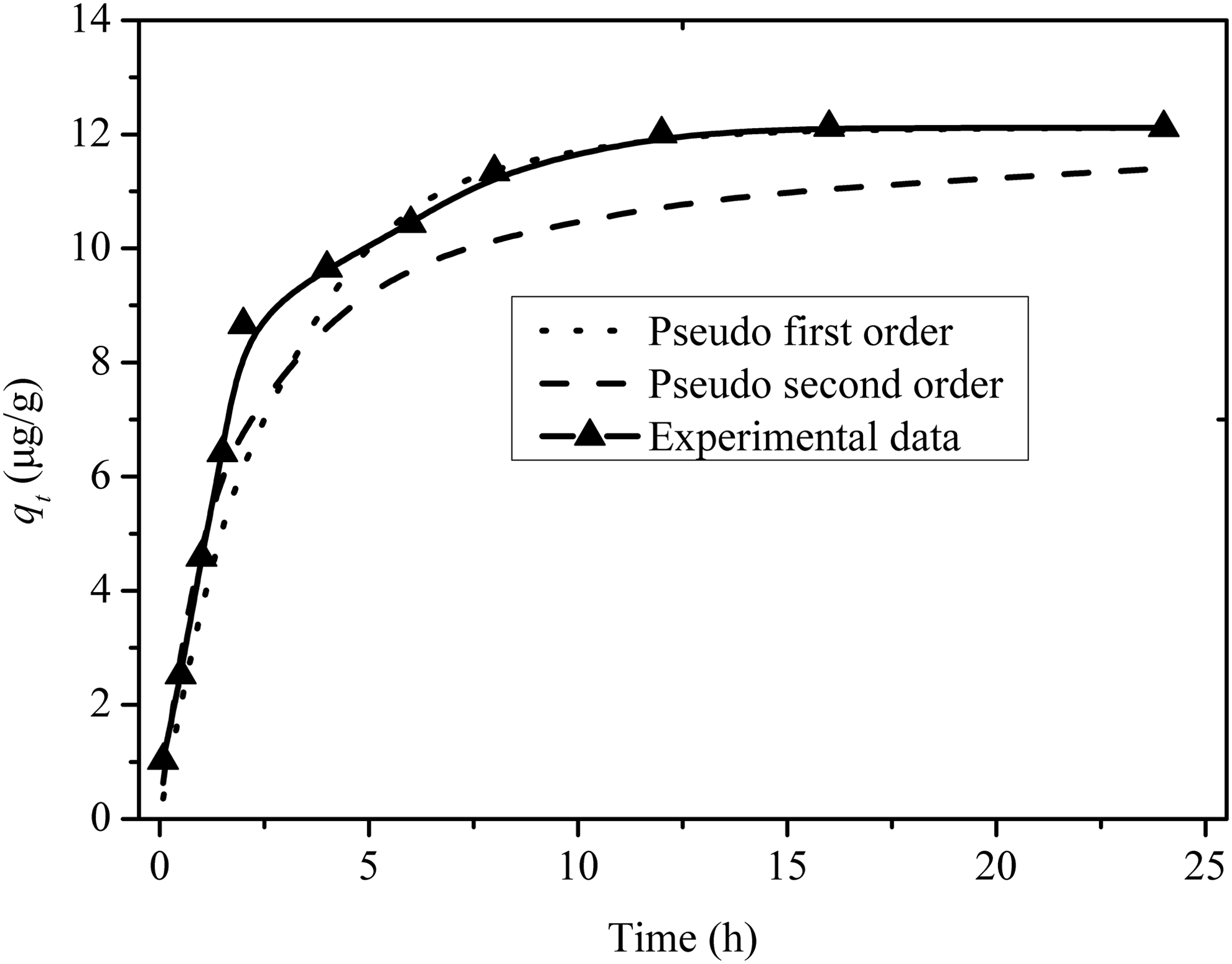
Pseudo-first-order and pseudo-second-order kinetic curves for PHE sorption on limestone. Conditions: sorbent dosage, 0.5 g; initial PHE concentration, 500 μg/L; temperature, 25°C; contact time, 24 h; pH 7.0. PHE, phenanthrene.
Conditions: sorbent dosage, 0.5 g; initial phenanthrene concentration, 500 μg/L; temperature, 25°C; contact time, 24 h; pH 7.0.
Generally, the intraparticle diffusion mechanism is one of the most limiting factors that control the sorption kinetics (Guibal et al., 2003). The intraparticle diffusion model can be described as follows (Lagergren, 1898):
Figure 5 plots qt versus t0.5 at different times, indicating that the plot of qt versus t0.5 shows a multilinear trend. The intraparticle diffusion constants were calculated using Equation (5). The values of intercept give an idea about the boundary layer thickness: the larger the intercept is, the greater the boundary layer effect (McKay et al., 1985). The boundary layer resistance will affect the rate of sorption and increase in contact time. When the plots do not pass through the origin, this is indicative of some degree of boundary layer control and further shows that the intraparticle diffusion is not the only rate-limiting step, but also other kinetics models may control the rate of sorption, all of which may be operating simultaneously. The sorption of PHE can be divided into three stages. For the initial concentrations, the first stage was completed within the first 2 h and the second stage of intraparticle diffusion control was then attained. The third stage only occurred after 12 h. The rates of different stages observed indicated that the adsorption rate was initially fast and then slowed down when the time increased. At the beginning, PHE was adsorbed by the exterior surface of limestone. When the sorption of the exterior surface reached saturation, the molecular PHE might enter intraparticle pores in the limestone and adsorbed by the interior surface of the particle. During the diffusion of the molecular PHE, diffusion resistance may increase, causing the observed decrease in the diffusion rate.
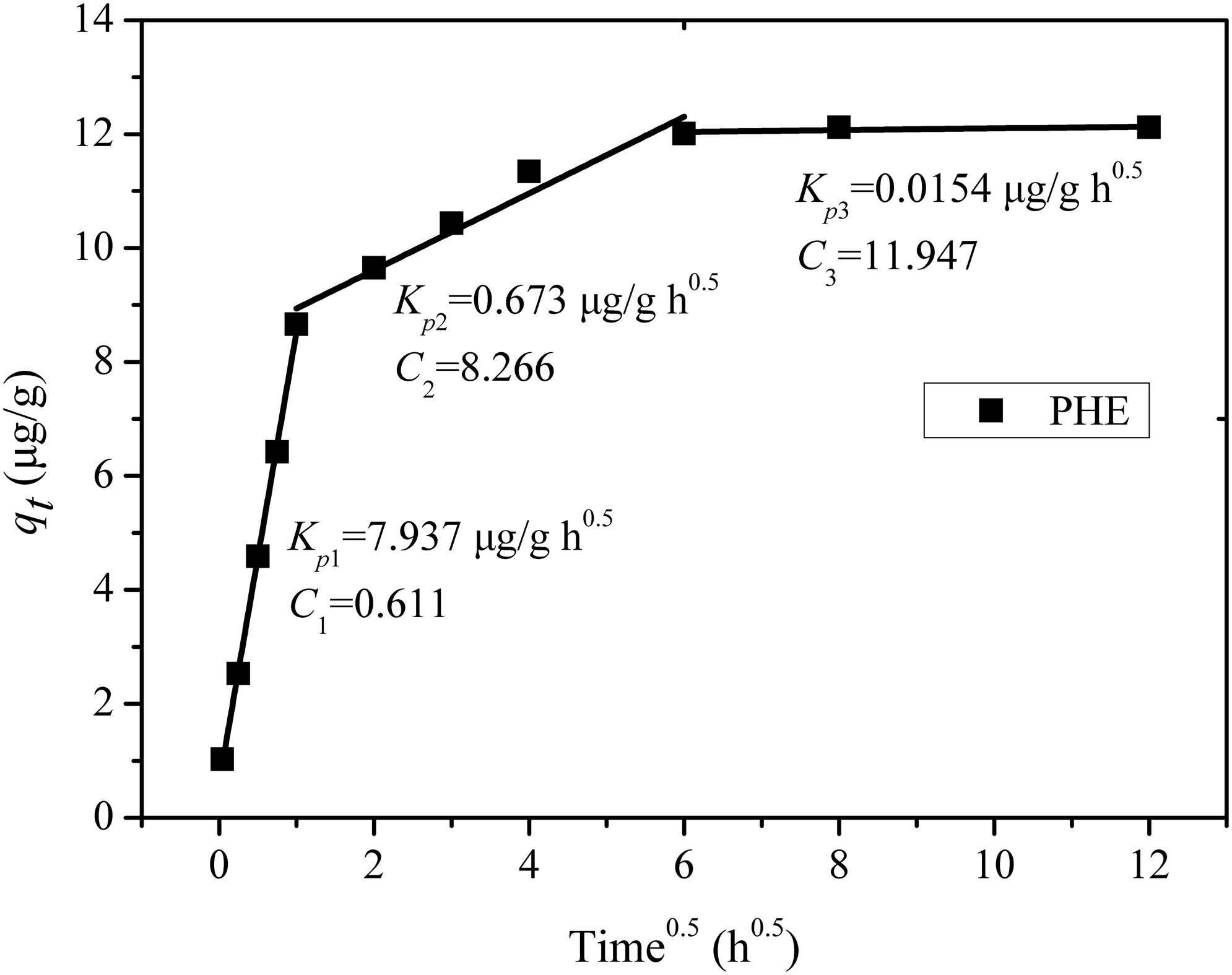
Linear regression of kinetic plot: intraparticle diffusion model. Conditions were as described in Fig. 4.
Generally, the last step is the equilibrium reaction and it is very rapid; the resistance is hence assumed to be negligible. The slowest step determines the rate-controlling parameter in the adsorption process. In our experiment, the rate of external transport was higher than the internal transport. So the adsorption of PHE on limestone was controlled by the particle diffusion.
Factors affecting PHE sorption on limestone
Carbonate and OC
To reveal the effect of carbonate and organic matter on PHE sorption, limestone sample was sequentially treated to remove carbonate and OC. Sorption experiments using S0, S1, and S2 fractions were then performed and the corresponding parameters are summarized in Table 2. The specific surface area was slightly increased from S0 to S1. Comparing the sorption isotherms of PHE from S0 to S2 and judging from the slight decrease of Kd value, carbonate played a minor role in the sorption, despite its dominant content in the limestone. A previous work with different results was done on river sediments by Wang et al. (2008) indicating that the sample was remarkably aggregated and came to exist with amorphous state after the removal of carbonate and that carbonate provided sorption sites in the PHE sorption process and thus the sorption mass decreased after removing carbonate in sediment samples.
Conditions: sorbent dosage, 0.5 g; phenanthrene concentration range, 50–500 μg/L; temperature, 25°C; contact time 12 h; pH 7.0.
TOC, total organic carbon.
Although the content of organic matter in limestone was only 0.33%, the contribution percentage of organic matter for PHE adsorption could be significant. The Kd value decreased from 87.89 to 22.45 L/g when OC was removed by H2O2 in our experiments. The organic matter in limestone may interact with organic contaminants through various binding and sorption interactions that considerably affect the migration and transformation of hydrophobic organic pollutants (Akkanen et al., 2012). There was a good linear correlation in a plot of Kd versus percent TOC, the value of R2 was 0.991 (Fig. 6b). And the decrease in Kd value can be compared to show that the contribution of OC was much higher than that of carbonate. Therefore, OC has a dominated effect on the sorption process. The R2 of linear isotherm decreased from 0.960 to 0.922 after removal of OC. The sorption of PAHs by inorganic fraction (i.e., carbonate, residue), represented by Freundlich isotherm, was primarily a physical process and the contaminants were adsorbed on many sorption sites. By contrast, via partition process, PAHs were partitioned into the organic phase, and the sorption isotherms present linear behavior (Hundal et al., 2001), which may explain why the linear absorption coefficient decreased from S0 to S2 samples in our experiments.

Effects of carbonate and organic carbon on PHE sorption.
pH
Experimental results of PHE sorption on limestone under different pH conditions are shown in Fig. 7. It can be seen that the sorption significantly decreased under both acidic and alkaline conditions.

Effects of pH on PHE sorption.
FTIR spectra results showed the existence of hydroxyl groups. The OH groups resulted in hydrophobic behavior of the limestone material. This information was very important when considering the use of this material as adsorbent of nonpolar organic compounds, like PAHs (Vidal et al., 2011). Most of the surface hydroxyl groups are at neutral state (Zhang et al., 2008); under acidic conditions, the hydroxyl groups are becoming protonated to OH2+ groups, resulting in the decrease of the sorption of PHE. And, acidic condition was thought to lead the dissolution of carbonate, which reduced the sorption sites for PHE.
In general, the Kd value decreased as pH increased from 7 to 12. Similar result was observed in previous studies that PAHs sorption capacity of soil/sediment decreased when the pH values increased (Ping et al., 2006; Yan et al., 2013). PAHs tend to associate with dissolved OC, which could strongly affect the sorption behavior (Shi et al., 2007). The CH2 signal at 1050 cm−1 decreased as pH increased, which indicated that OC, as a source of organic CH2 groups, desorbed from the surface at alkaline condition. Also, the decrease of PHE sorption could be attributed to the change of the carboxy groups on the surface of limestone. The higher the pH was, the greater would be the dissociation of the functional groups, thus affecting the sorption performance of the adsorbent (Zhou et al., 2012).
Ionic strength
The results of ionic strength effects using different concentration of CaCl2 (varied from 0 to 0.2 M) for PHE sorption are shown in Fig. 8. The Kd and qe values were found to decrease with change of CaCl2 from 0 to 0.005 M. Few ions in the solution possibly resulted in increase of diffuse double layer thickness and absolute value of zeta potential on the surfaces of limestone particles (Rao and He, 2006). It could be correspondingly difficult for PHE to approach and adsorb on limestone. A pore-filling mechanism was suggested by Xia and Ball (1999) to explain PAHs sorption within a microporous solid: when there was a small amount of calcium ions, some pores would have been occupied. This might be a reason for the slight decrease of the sorption amount of PHE on limestone with addition of CaCl2.

Effects of ionic strength on PHE sorption: sorption isotherms of PHE
On the contrary, Kd and qe values increased with the increase of CaCl2 concentration greater than 0.005 M. A possible reason for this is that adding CaCl2 to the solution may have a salt-out effect on the sorption process similar to that of partitioning a nonionic compound into two phases, one of them being water (Qian et al., 2011). So, addition of calcium salt may decrease the solubility of PHE in the aqueous solution and increase its partition into the solid phase. Dias et al. (2013) arrived at the same conclusion: increase of the ionic strength, which further reduced the solubility of PAHs in water, resulted in precipitation or adsorption onto the glass walls. Calcium cations also cause organic matter to form aggregates, which could create a new solid “phase” for additional sorption of PHE. Meanwhile, calcium ions can form a cation bridging between PHE and the surface of the limestone.
The effect of CaCl2 concentration on the sorption process can be quantitatively described using the following equation (El-Nahhal and Safi, 2004):
where Cw is the amount of PHE adsorbed from the distilled water, Cs is the amount of PHE adsorbed at a certain calcium chloride concentration of water, [CaCl2] is the concentration of CaCl2, and K is the salt-out constant.
Our experiment results showed a good fit of Equation (6). The mean value of K was 0.81 and the relative standard deviation was 6.2%.
Further, the changes of the molar free energy (ΔG0, 25°C) of PHE sorption can be calculated using Equation (7) (El-Nahhal and Safi, 2004):
where ΔG0 is the molar free energy change (kcal/mol1), R is the gas constant (1.986 cal/[K·mol]), and T is the absolute temperature.
Molar free energy values of PHE sorption decreased from −4.78 to −5.15 kcal mol−1 when the CaCl2 concentration increased from 0.005 to 0.1 M. These data provided additional evidence that the sorption of PHE on limestone should be mainly a physical process. And the value of ΔG0 was reduced as CaCl2 concentration increased. Thus, sorption of PHE should be a spontaneous reaction and salt-out effects will lead to the increase of sorption capacity.
However, when the concentration of CaCl2 was higher than 0.1 M, the qe values remained constant, which could not be explained by the “salting-out”, or cation bridge. Strong interactions can occur between aromatic π donors and metal cations in aqueous solutions, which may also be relevant (Keiluweit and Kleber, 2009).
Humic acids
To understand the interaction of HA and PHE sorption on limestone, three sets of experiments were designed and conducted as follows: (1) 0.5 g limestone mixed with 10 mg/L HA was placed on a shaker to equilibrate; after 12 h, 300 μg/L PHE was added into the solution. (2) 0.5 g limestone mixed with 300 μg/L PHE was placed on a shaker to equilibrate; after 12 h, 10 mg/L HA was added into the solution. (3) HA and PHE were added at the same time.
After PHE was added into the equilibrium solution of HA, it was observed that the HA concentration in solution did not obviously increase but the PHE concentration sharply decreased (Fig. 9a, c). This phenomenon indicated that there were still some pores for PHE after the HA adsorption equilibrium. And there was a competition adsorption between PHE and HA, the affinity of HA and limestone was stronger than that of PHE. So PHE can only replace a small amount of HA. Therefore, the concentration of HA in solution did not obviously increase. By contrast, after the HA was added into the equilibrium solution of PHE, the concentration of PHE sharply increased (Fig. 9d). Thus, competitiveness of HA on sorption sites of limestone was stronger than PHE, and HA can desorb PHE from the limestone.

Effects of humid acid on PHE sorption:
Sorption parameters of PHE (added at the same time with HA) were determined in the presence of HA at concentrations from 0 to 100 mg/L. The Kd and qe values increased first and decreased thereafter with the increase of added HA concentrations (Fig. 9e, f). Cumulative adsorption may develop between exogenous HA and PHE when the concentration of HA is lower (Totsche et al., 1997). Once HA was adsorbed on the limestone, the OC content of the adsorbent was elevated, the adsorbed HA on solids had far stronger capacity of PHE uptake than the original organic matter (Gao et al., 2007), thus promoting PHE sorption on limestone.
However, in the range of HA concentrations we tested, the Kd and qe values turned out to decrease after the initial increase. This could be the result of the enhanced PHE association with HA, which increased the solubility of PHE in solution, reducing the sorbability of the limestone. The binding constant between HA and PHE was greater than the sorption constant Kd (Gauthier et al., 1986), with the continuous increase of the HA concentration. When the combination between HA and PHE was enhanced in the aqueous solution, HA would adsorb PHE and competitive sorption between HA and limestone occur.
ACE
From the kinetics curves of PHE and ACE sorption on limestone (Fig. 10), it can be seen that the sorption rate of PHE was faster than that of ACE, and therefore PHE would be adsorbed on the limestone surface preferentially. Constant initial concentration of the first solute (PHE or ACE) but constantly varying concentrations of second solute (ACE or PHE) were employed in these experiments. An increase in concentration of PHE from 0 to 40 μg/L was accompanied by an increase in ACE sorption. As noted in earlier studies, the high molecular weight PHE was adsorbed on the limestone surface preferentially, which resulted in increase of organic matter content (McGinley et al., 1993). Thus, the qe increased from 8.03 to 8.43 μg/g. However, this pattern of behavior was opposite when the concentration of PHE was greater than 40 μg/L. And as shown in Fig. 11, increase in ACE concentration was accompanied by reduction of PHE sorption. The observed reductions in PHE sorption on limestone could be attributed to the competitive sorption between PHE and ACE at elevated concentrations of ACE.
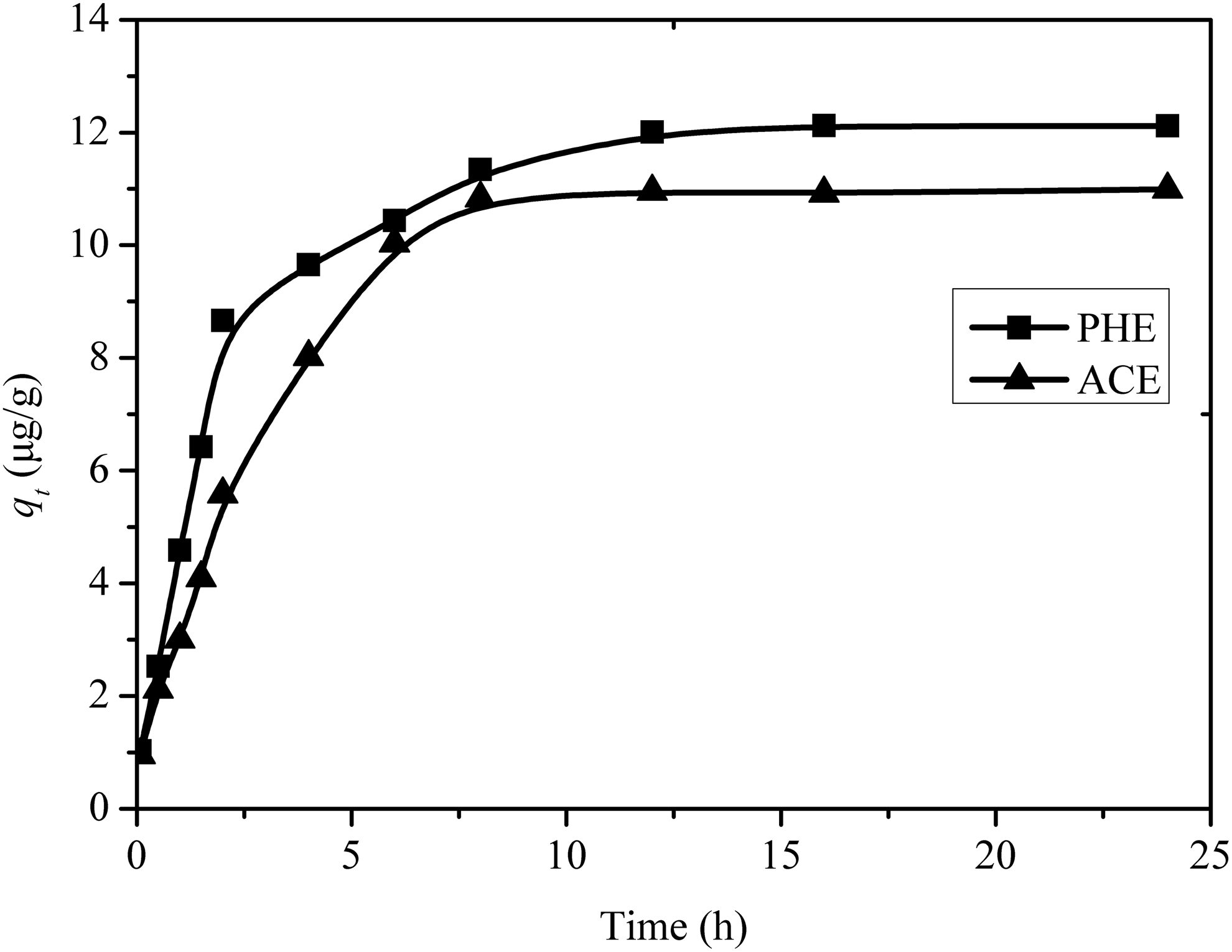
Kinetic curves of PHE and ACE sorption on limestone. Conditions were as described in Fig. 4, with initial ACE concentration of 200 μg/L. ACE, acenaphthylene.
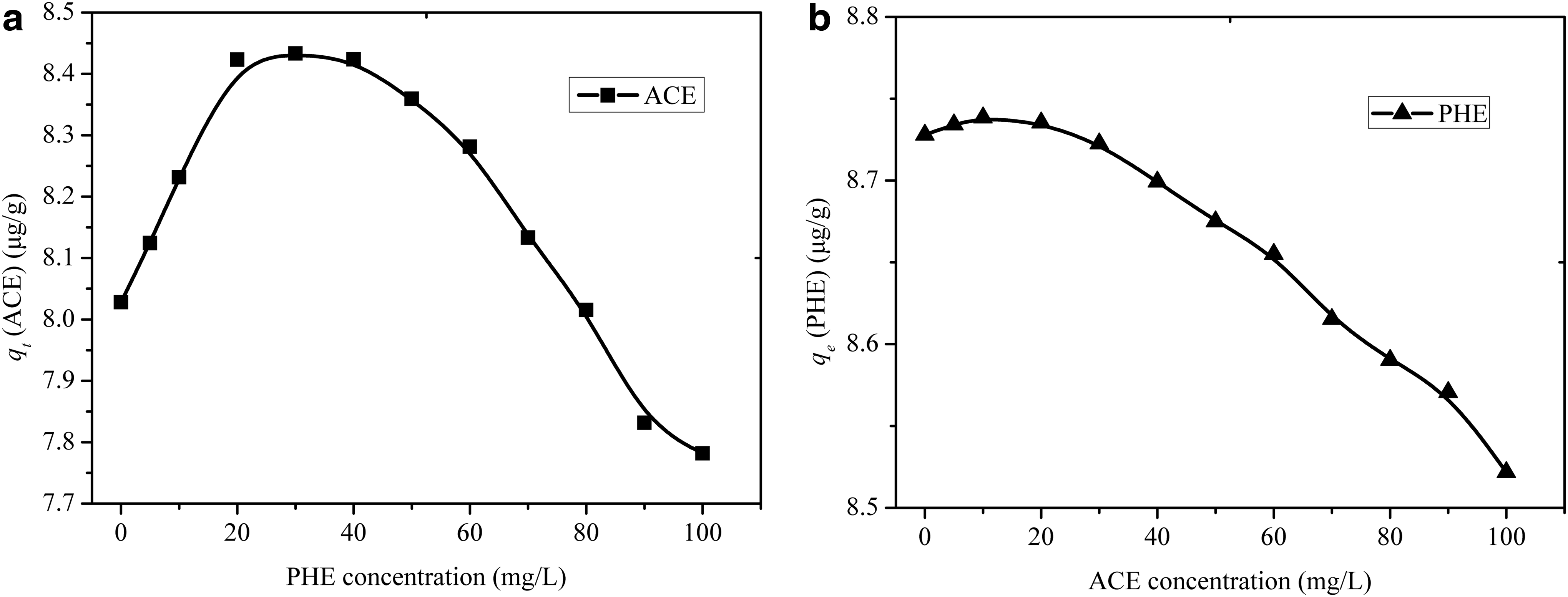
The qe value at different coexistence conditions:
Conclusions
Adsorption kinetics and controlling factors of PHE were studied in a batch experimental system. The following results were obtained:
1. PHE adsorption behaviour was described by a liner isotherm. The adsorption process of PHE on limestone could be well described by intraparticle diffusion model, and the adsorption rate was mainly controlled by the diffusion rate of the molecular PHE within a particle. 2. Because carbonate affected physical characteristics of limestone, it exerted slight influence on the adsorption behavior of PHE. On the other hand, due to the fact that organic matter mainly determined partition procedure, its effect on adsorption of PHE was remarkable. 3. Several factors that affect PHE adsorption on limestone were investigated in this study. The adsorption significantly decreased under both acidic and alkaline conditions. Addition of CaCl2 enhanced the adsorption due to salt-out effect. 4. In the presence of PHE in HA sorption equilibrium solution, the affinity of HA and limestone was stronger than that of PHE. PHE can only replace a small amount of HA due to the stronger affinity of limestone and HA. However, after the HA was added into the equilibrium solution of PHE, it can desorb PHE from the limestone.
Footnotes
Acknowledgments
This research work was financially supported by National Natural Science Foundation of China (No. 40902071, and No. 41120124003), the Ministry of Science and Technology of China (2012AA062602), and the Ministry of Education of China (111 project and Priority Development Projects of SRFDP [20120145130001]).
Author Disclosure Statement
No competing financial interests exist.