
Abstract
Abstract
Hydrogen sulfide (H2S) is commonly detected among waste gases from wastewater treatment plants causing atmospheric pollution, which affects human and environmental health. The aim of this study is to characterize the microbial diversity present within two different H2S biofiltration systems packed with either pozzolan or marble and to assess their efficiency for H2S removal. Few examples were reported about the relationship between the characteristics of the packing material and biofilter performance. Physicochemical parameters such as sulfate concentrations and pH are measured along this study. Sulfate concentrations produced by H2S oxidation were higher using marble biofilter and a higher efficiency of H2S removal was observed. Microbial diversity was characterized using a culture-independent 16S rRNA gene approach. Our results show that the biofilter packed with marble is very acidic (pH <3) and exhibits a higher bacterial diversity with three dominant bacterial families: Xanthomonadaceae, Hydrogenophilaceae, and Spirillaceae. However the biofilter packed with pozzolan is less acidic (pH 5.7–6.8) and shows completely different phylotypes where the plastid 16S rRNA gene of the red algae Cyanidium caldarium and Acidithiobacillus bacterium were retrieved. Such results show that the selection of an appropriate packing material is essential to the odor removal performance of a biofilter system and should allow for the design of better reactor with optimal operating conditions.
Introduction
O
Materials and Methods
Biofiltration system description
Different biofitration systems were set up at the WWTP of North Hammam-Sousse, in Tunisia (sized to treat 17,500 m3/day of waste water, 4,750 kg BOD5, 132,000 population equivalents). The wastewater discharged from the WWTP is characterized by the presence of H2S concentration >500 ppm. The inorganic packing material of the biofilter used in this study is either the pozzolan to which a calcareous material is added in double layer, or a marble. Carbon sources needed for bacterial growth are carbonates brought by the water and the CO2 conveyed by the gas to be treated. The use of mineral supports with regular size grading allows sizing these biofilters with a flow rate ranging from 500 to 750 m3/h. All biofiltration systems are operating at a constant atmospheric mesophile temperature (30°C±1°C).
The biofiltration system using marble as support treats the used air of the primary decanter, and the other which uses pozzolan, treats the used air of the stabilizer. The gas was pumped by an air blower and then passed through the biofilter bed. Each biofilter is watered during 10–15 min/h, with uncluttered water filtered to remove suspension materials.
Sludge sampling
Each tested biofilter was sampled in June 2007, and 1.5 L was collected. Samples were taken from the upper and the middle bed of the biofilter system, and washed three times with phosphate buffered saline (PBS) by shacking vigorously when adding PBS to dissociate microbes stacked on pozzolan or marble gravels. Samples were then pelleted by centrifugation at 20,000g for 15 min at 4°C and frozen at −20°C for further analysis.
DNA extraction, PCR amplification of 16S rRNA gene, cloning, and sequencing
Genomic DNA was extracted from 100 to 200 mg aliquots as described previously (Chouari et al., 2003). Polymerase chain reaction (PCR) amplification and cloning were carried out as already described with specific primers for Bacteria (008F—AGAGTTTGATCCTGGCTCAG) (Hicks et al., 1992) or (063F—CAGGCCTAACACATGCAAGTC) (Marchesi et al., 1998) domains and a reverse universal primer (1390R—GACGGGCGGTGTGTAC AA) (Zheng et al., 1996). The nucleotide sequence of plasmid inserts was determined by classical automated Sanger sequencing (3730XL sequencer; Applied Biosystems).
The comparison of the obtained 16S rRNA gene sequences to those present in databases, through computing tools, allows establishing a phylogenetic analysis and identification of organisms by passing the cultural methods, which introduce numerous bias to the real bacterial diversity present within the sample.
Sequence analyses
The 16S rRNA gene sequences were assembled using Phrap (www.phrap.org). Only good quality 16S rRNA gene sequences (longer than 1,000 bp and with Phred qualities of above 15 for each base) were selected for further analysis. Chimera sequences were searched before analysis by using the procedure described by Juretschko et al. (1998). Libraries were simultaneously called AB and AC for the first sample issued from the marble biofilter treating used air emanating from the primary settling basin and AD and AE for the second sample issued from a biofilter of pozzolan treating used air emanating from the stabilizer.
Phylogenetic affiliation
The retrieved sequences were compared by basic local alignment search tool (BLAST) (Altschul et al., 1990) to two different databases: the Greengenes database (DeSantis et al., 2006) and the Ribosomal Database Project 9.50 (Maidak et al., 1994). Sequences were phylogenetically affiliated with a given taxon only if the alignment length was 1,000 bp with 90% sequence identity. Otherwise, sequences were considered unclassified if these criteria were not respected or if the results from the different databases were different.
Operational taxonomic unit assignment
16S rRNA gene sequences were aligned with MAFFT (Katoh et al., 2002) and operational taxonomic units (OTUs) were defined using the furthest neighbor clustering algorithm of distance-based OTU and richness (DOTUR) software with a 97% identity threshold (Strackebrandt and Goebel, 1994; Schloss and Handelsman, 2005). Diversity coverage was calculated using the Good's formula (Good, 1953).
Statistical comparison of the clone libraries
The four libraries were compared bilaterally with R-Libshuff (Schloss et al., 2004) using the distance matrix calculated by DNAdist as input. Using a Monte Carlo procedure, the probabilities that the observed difference among the digesters is due to chance were calculated. With an experiment-wise error rate of 0.05, and taking into account a Bonferroni correction due to the multiple comparisons, the libraries were considered significantly different if the p-value is less than 0.000712 (Schloss et al., 2004).
Shared OTUs and similarity
Results obtained with DOTUR at a distance of 0.03 were used to run shared OTUs and similarity (SONS) software (Schloss and Handelsman, 2006) and to compare the four 16S clone libraries. SONS was used to evaluate the membership and communities structure by accounting for OTU abundance and distributions that are either specific to one library or shared by two libraries. By comparing the four libraries, SONS allowed us to distinguish the shared OTUs among the total number of observed OTUs and the number of sequences in each OTU.
Nucleotide sequence accession numbers
Sequences of OTUs reported in this study were deposited in the EMBL databases under accession numbers from KJ756848 to KJ757030 and from KJ707249 and KJ707453. The correspondence among accession numbers, clones names, and OTU numbers is given in Supplementary Table S1.
Physicochemical analyses
The pH and sulfate concentrations were measured daily during the 3 preceding months of sampling. The pH was measured using a digital calibrated pH-meter (HANNA pH 210). Sulfate concentrations were determined using the barium protocol according to the procedure listed in APHA (1998). Gas samples were collected in Tedlar bags. Inlet and outlet H2S gas concentrations were measured weekly in triplicates by gas detector tubes (Rohrchen). The detection ranges of the H2S gas tubes were 1,000–3,600 ppm and 10–630 ppm for inlet and outlet H2S gas concentrations, respectively. The removal efficiency was determined as presented in the Equation (1):
RE, removal efficiency (in %); Cin: inlet H2S concentration; Cout: outlet H2S concentration.
Results
The diversity of bacterial communities present in two different packaging biofilters of the same WWTP treating used air were studied using comparative sequence analyses of 16S rRNA genes. According to phylogenetic analyses established by BLAST, all 16S rRNA gene sequences fell into nine lineages of Bacteria, one candidate division, and some unknown bacteria.
Phylogenetic analyses of 16S rRNA gene of AB and AC libraries (marble biofilter)
The present molecular analysis shows that within the 581 obtained 16S rRNA gene sequences, 197 sequences retrieved from AB clone library, and 220 from AC and 165 sequences are commonly found in both clone libraries. The bacterial sequences were grouped into OTUs using a 97% sequence similarity cut-off (Stackebrandt and Goebel, 1994). According to this definition, 372 OTUs were defined and distributed as follows: 16 for AB, 181 for AC library, and 27 OTUs were common to both libraries (Table 1). BLAST analysis shows 202 OTUs were affiliated—with more than 97% similarity—with cultivable (57 OTUs in total) or not-yet-cultivable representative microorganisms (145 OTUs in total), whereas 170 OTUs (45.7%) are representing novel phylotypes.
[1− (n/N)]×100 (Good, 1953).
Richness estimations: values in parentheses are the 95% confidence intervals.
OTU, operational taxonomic unit.
BLAST analyses shows that in both AB and AC libraries, Proteobacteria and Bacteroidetes are the most dominant phylogenetic groups representing 64.3% and 11% of the total OTUs, respectively. The remaining 11.8% of the total OTUs are affiliated with Firmicutes, Chloroflexi, Gemmatinomonadetes, Cyanobacteria, Actinobacteria, Plantomycetes, Acidobacteria, and NKB-19 candidate division with ≤3% for each phylum. 12.9% of the sequences are affiliated with unclassified bacteria (Table 2).
97% similarity sequence to the most related sequence having a cultivable representative.
≥97% similarity sequence to the most related sequence with no cultivable representative.
<97% similarity sequence to any known sequence.
Fifty percent of the Proteobacteria affiliated OTUs (52% and 51% for AB and AC library, respectively) represent novel phylotypes showing ≤97% sequence similarity with the most related 16S rRNA gene sequence present in the Genbank database. Three OTUs are predominant within all analyzed clone sequences. The first predominant OTU (5.9% of all sequences; 34/581 sequences) is affiliated with an uncultured Spirillaceae (EF467563), a Betaproteobacterium, isolated from Frasassi sulfidic cave stream biofilm in Italy, with ≥99% similarity. The second well-represented OTU is affiliated with Thiobacillus sajanensis sp. nov., (with >98% similarity), a new obligate autotrophic sulfur-oxidizing bacterium isolated from Khoito-Gol hydrogen sulfide springs in the Eastern Sayan Mountains (Buryatia). This Betaproteobacterium represents 5% (29/581 sequences) of the total sequences. The third predominant OTU represents 3.3% (19/581 sequences) of all sequences is affiliated with Lysobacter (EU273938) (with ≥97% of sequence similarity), a Xanthomonadacea of the Gammaproteobacteria isolated in South Korea from a soil of ginseng field.
AB and AC clone libraries profile is quite qualitatively and quantitatively the same in terms of retrieved lineages, except that Gemmatinomonadetes, Betaproteobacteria, and Gammaproteobacteria are more detected in the AB library, whereas Alphaproteobacteria are more retrieved in the AC library. Planctomyces and NKB-19 candidate division are only detected in the AB library.
Phylogenetic analyses of 16S rRNA gene of AD and AE libraries (pozzolan biofilter)
A total of 507 16S rRNA gene sequences are analyzed. Most of them (481 sequences which represent 94.9% of all sequences) were retrieved within the two libraries (Table 3). Only 22 and 4 sequences for AD and AE libraries, respectively are specific of each constructed library. A poor diversity was observed in this biofilter sample as all the sequences were grouped into only 16 OTUs. Indeed, all the OTUs show ≥97% sequence similarity with a cultivable representative (for 1 OTU) or a noncultivable one (for 15 OTUs). This low diversity is representative of the studied sample as coverage estimates indicated at least 95% for each library. Analyses at the phylogenetic group level (Table 4) shows that Gammaproteobacteria is the predominant group with 50% of all OTUs in AD and AE libraries, followed by Cyanidium caldarium 16S rRNA plastid gene (25.0% and 33.3% of all OTUs in AD and AE libraries, respectively) and unclassified bacteria (8.3% and 16.7%). Betaproteobacteria and Actinobacteria represent 8.3% of all defined OTUs in the AD library, but they were not detected in the AE library. Two OTUs were predominant in both AD and AE libraries. One OTU is affiliated with a plastid 16S rRNA gene of C. Caldarium cloned sequence (EF613020.1) (Maid and Zetsche, 1990) with ≥99% sequence similarity and represents 51.1% and 45.5% of all sequences retrieved from AD and AE libraries, respectively. The second most dominant OTU is affiliated with Acidithiobacillus with ≥99% sequence similarity, a Gammaproteobacteria cloned sequence (DQ499168.1) (Macalady et al., 2007), which was recently proposed as a new phylum (Acidithiobacillia) (Williams et al., 2013), and represents 28.6% and 38.4% of all analyzed sequences of AD and AE libraries, respectively.
[1− (n/N)]×100 (Good, 1953).
Richness estimations: values in parentheses are the 95% confidence intervals.
AD and AE clone libraries profile is quite qualitatively and quantitatively the same in terms of defined phylotypes, except for one OTU (AB255113.1), which represents 8.3% of all OTUs of the AD library, and is affiliated with the Actinobacteria.
Evaluation of diversity coverage and richness of the clone libraries and comparison of bacterial communities' diversity within the two analyzed samples
To evaluate the level of information contained in the different libraries, several parameters were measured. First we calculated Shannon index to take into account the evenness of the OTUs. An important value for AB and AC libraries in comparison to AD and AE libraries were obtained (Tables 1 and Table 3). This confirms that the marble biofilter sample shows a higher diversity in comparison with the pozzolan biofilter. Second, we compared the observed number of OTUs to the estimated total number of OTUs. We used two estimators, ACE and Chao1, to calculate the total diversity of each sample based on the observed OTUs. The observed number of bacterial OTUs is far from reaching the estimated total number of OTUs for AB and AC libraries. This shows that the diversity was not exhaustively sampled. However, the observed number of OTUs obtained for AD and AE libraries approaches the total diversity of the sample given by the estimators. This result indicates that our libraries cover almost all the diversity originally present in the sample. Nevertheless, taking into account the confidence interval which is large, this indicates that the estimation is less accurate and is due to the proportion of one sequence containing OTUs, consequently, the value of the estimators increases. Another method for evaluating the level of information contained in the libraries is the calculation of the diversity coverage, which represents the percentage of a chance for a clone newly sequenced to belong to an OTU already observed. In this study, diversity coverage values are 47.1%, 46%, 95.5%, and 95% for AB, AC, AD, and AE libraries, respectively. These results show that for AB and AC libraries do not reflect the total diversity of the original sample due to the large diversity encountered in this environment. Comparisons of the different communities will help determine which proportion of the population is shared with the other and which proportion is specific of the different libraries.
The∫-Libshuff software was used to compare the libraries sequence by sequence for a range of distances taking into account both the frequently observed and rare sequences. A pairwise comparison was achieved for the four libraries using the∫-Libshuff, but the interest of this comparison is essentially the difference between the two samples: AB-AC libraries versus AD-AE libraries. With 372 OTUs for the first sample (AB and AC libraries) versus 16 OTUs for the second sample (AD and AE libraries), bacterial diversity is quite different. Even if the Gammaproteobacteria are the most represented organisms in both samples, there is no shared OTU between the different samples. Moreover, 16S rRNA plastid gene of C. caldarium, the second most abundant phylogenetic group in AD and AE libraries, was not detected in AB and AC libraries. Bacteroidetes was also not retrieved within the second sample.
Physicochemical analyses of the tested biofiltration systems
Microbial metabolism depends strongly on temperature and pH (Ma et al., 2006). Biofiltration systems used in this study are closed structures designed to maintain a mesophile temperature (30°C±1°C). The pH was measured daily in the two tested biofilters during 3 months. Average measurements for each month are given in Fig. 1. A constant pH (between 7.59 and 7.74) characterizes the water of watering. Very acidic values ranging from 1.77 and 3.74 are obtained in the marble biofilter whereas slightly acidic to neutral values (from 5.70 to 6.81) have been reported in the pozzolan filter. Compared to the concentration of SO42− ions brought by the water, we noticed approximately three times and two times more SO42− in the percolate of marble and pozzolan biofilters, respectively. In fact, these concentration ranges are 889.6–1,218.7, 3,183.5–3,775.6, and 2,103.6–2,334.3 mg/L for the water of watering, marble percolate and pozzolan percolate, respectively (Fig. 2). Removal efficiency of H2S reaches ∼90–99% and 82.5–90% in percolate from marble and pozzolan biofilters, respectively (Fig. 3).
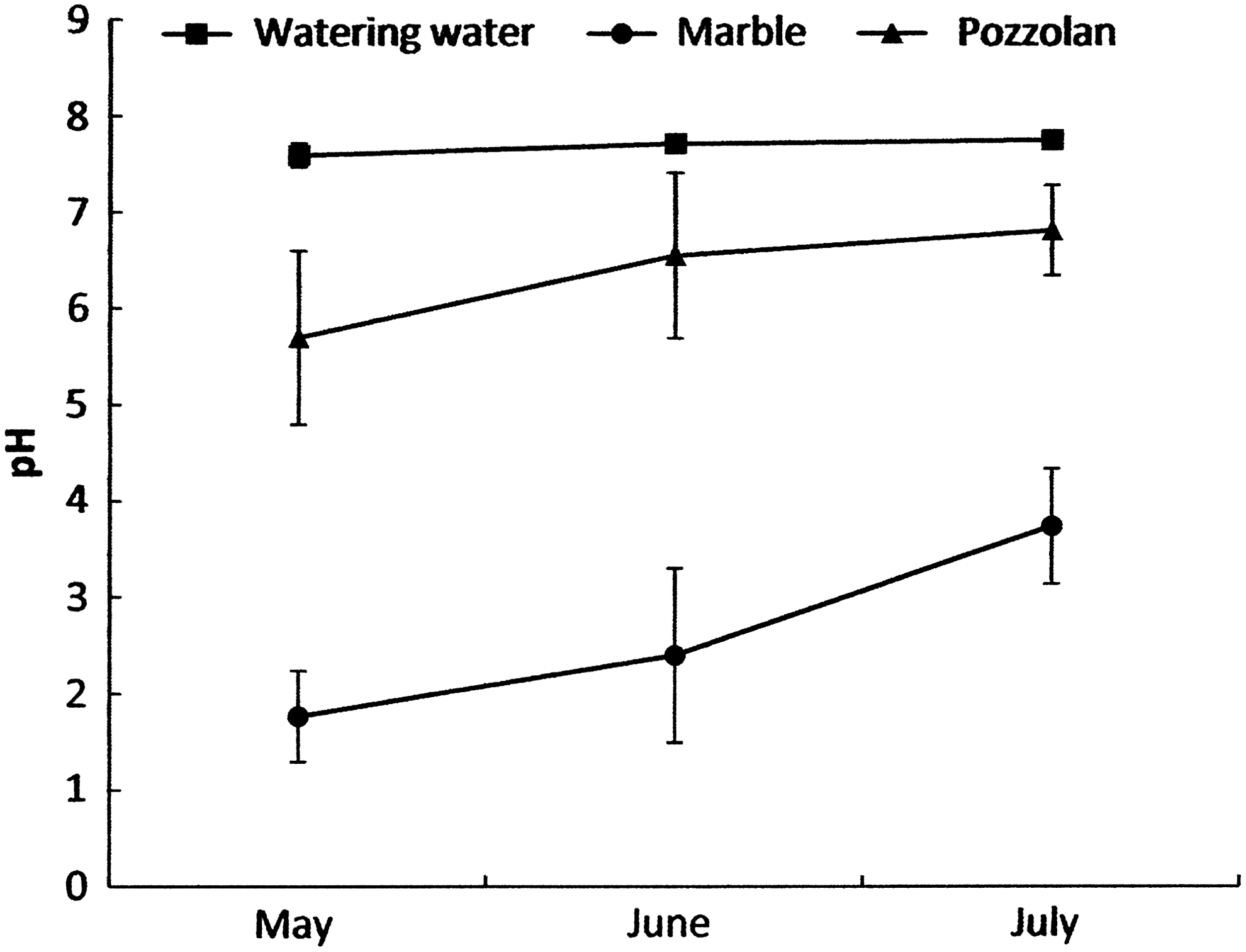
Comparison of pH values (average±error of daily measurements during 3 months: from May to July) in the watering water, in the percolate of the biofiltration treating system used air from the primary decanter (marble), and from the percolate of the biofiltration system treating used air from the stabilizer (pozzolan).
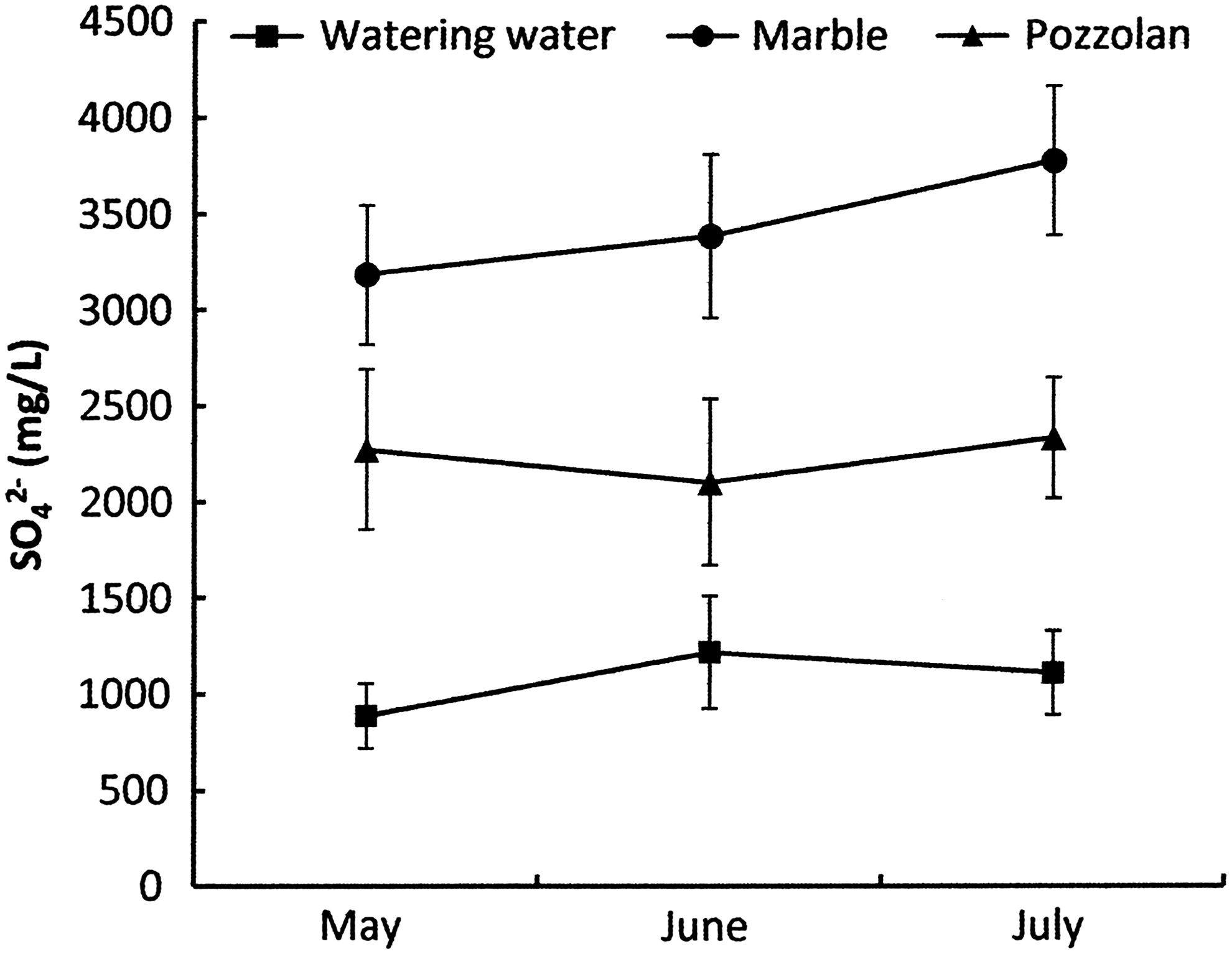
Comparison of SO42− (mg/L) production (average±standard error of daily measurements during 3 months: from May to July) in water, in the percolate of the biofiltration system treating used air from the primary decanter (marble), and from the percolate of the biofiltration treating system used air from the stabilizer (pozzolan).
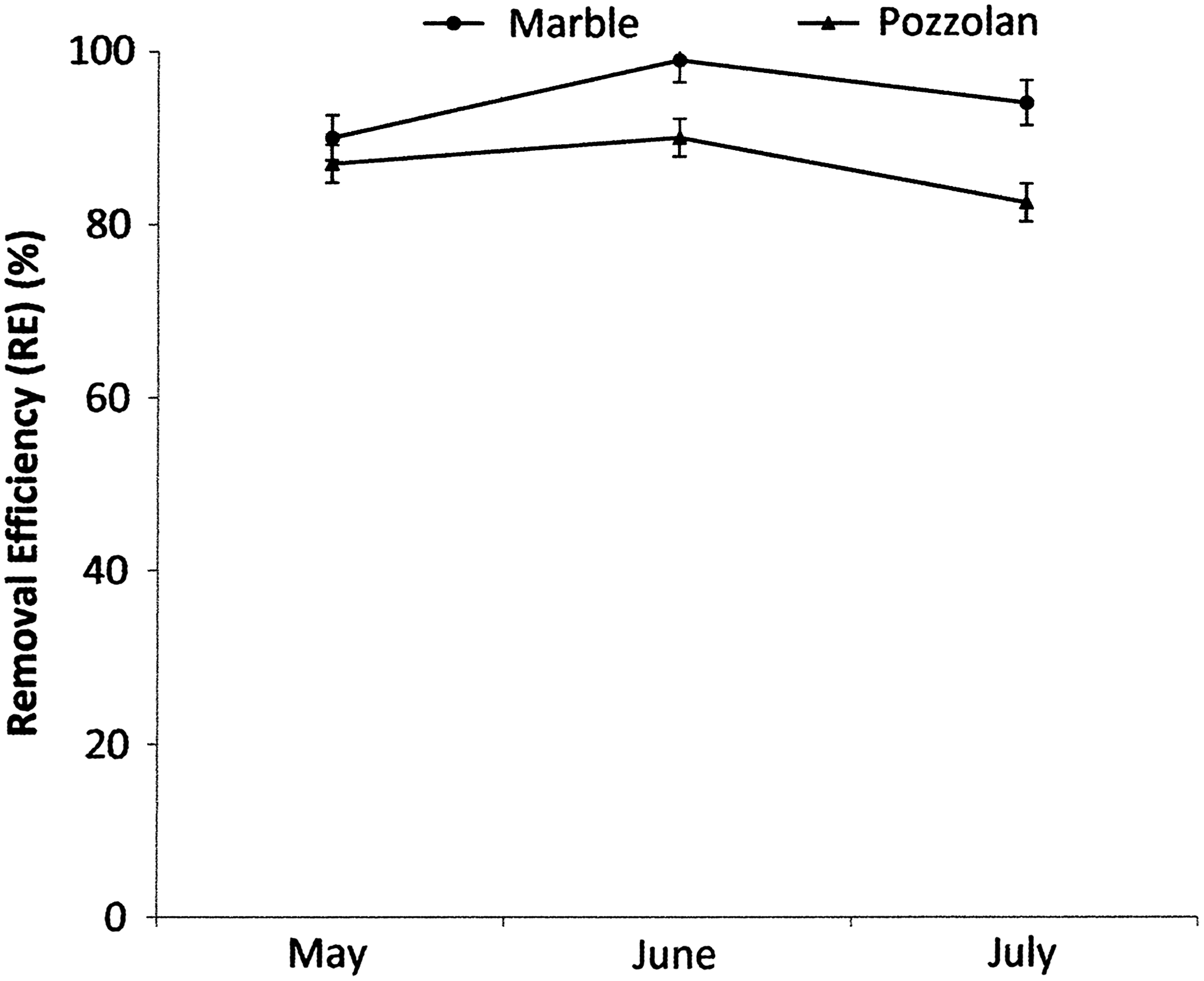
Hydrogen sulfide (H2S) removal efficiency in pozzolan and marble biofilters during operational period of 3 months (average±standard error of weekly measurements of H2S inlet and outlet concentrations).
Discussion
The objectives of this study are (1) to compare the bacterial diversity present in two different biofilter supports installed in a WWTP, (2) to identify potential microorganisms responsible for H2S removal, and (3) to study the eventual influence of the packing material on the biofilter's performance for H2S removal. As showed in previous studies (Duan et al., 2006; Omri et al., 2011), we found that low pH provides favorable conditions for H2S oxidation. In fact, photoautotrophic and chemolithotrophic sulfur-oxidizing bacteria (SOB) oxidizes H2S to sulfur ions or sulfate. Photoautotrophs use CO2 as the terminal electron acceptor, whereas chemolithotrophs use oxygen (aerobic species), nitrate, or nitrite (anaerobic species) as terminal electron acceptors (Tang et al., 2009). Sulfate and H+, products of microbial oxidation of H2S, are responsible for pH decrease, and even if active, the bacteria responsible for H2S removal have an optimum pH growth of 3 (Yang and Allen, 1994), they can grow in a wide range of pH like Thiobacillus. Our study pinpoints specific bacterial phylotypes potentially responsible for H2S removal by oxidation in two different biofilter support. As the only external source of bacteria was brought by the water flux within the biofilters, the comparison of microbial diversity present after H2S treatment in each biofilter shows that, for the marble biofilter, the main retrieved OTUs are affiliated with a Lysobacter (Xanthomonadacea) and a Thiobacillus sequence (Betaproteobacteria) and an uncultured Betaproteobacteria retrieved from an acidic, oxic, and sulfur-rich environment (Macalady et al., 2007). For pozzolan biofilter support, the main OTUs were affiliated with Acidithiobacillus and a 16S rRNA plastid gene sequence of the red alga C. caldarium. These sequences were previously retrieved in such environments by other authors (Yin et al., 2008; Charnnok et al., 2013). All these affiliated microorganisms are autotrophic and were already described as active members of H2S removal (Cho et al., 1992; Ding et al., 2006; Ramirez et al., 2011). Besides these predominant species, other less abundant bacteria were identified like Bacteroides, Alphaproteobacteria within the marble biofilters, and Actinobacteria within the pozzolan biofilter. In their previous study, Okabe et al. (2007) shows that these microorganisms were associated with SOB and were presumed to be responsible for concrete corrosion in sewer systems.
Our results show a difference in the bacterial species, depending on the nature of the packing material of the biofiltration system, which probably affect the conversion's rate of H2S to sulfates. While Omri et al. (2011) rather identified Pseudomonas, Moraxellacea, and Acinetobacter in biofilters packed with peat, Xie et al. (2009) identified Bacillus and Pseudomonas as active H2S removal in biofilters packed with waste straw and cortex. Thiobacillus, Sulfobacillus, and Alicyclobacillus were retrieved from packed compost as biofilter in the study of Ding et al. (2006). Ralebitso-Senior et al. (2012) showed that for all species support medium attachment is critical. Hence, Duan et al. (2005; 2006) showed that the Biological Activated Carbon operated biofilter showed a better performance than the virgin carbon one. Similarly, we show that the biofilter packed with marble gives a better performance of H2S removal than the one packed with pozzolan. Marble seems to be a better microorganism carrier in biofiltration. The biofilm formation might be affected by H2S adsorption capacity and adsorption rate of the packing material. A comparative study of different packing materials showing variations in H2S removal efficiency (Dumont et al., 2010) is in accordance with our work.
In most studies, H2S removal efficiency was achieved at the bioreactor or pilot scale. In the present work, we analyzed the diversity of oxidizing bacteria at the scale of the treatment plant at the industrial scale, which allows us to have a better view of the active and well-adapted microbial populations. Even if there is a core of bacteria known to be really active in H2S removal, such as Thiobacillus and Acidithiobacillus, the presence of associated bacteria may contribute to improve the treatment efficiency since we observe that SO42− concentrations are higher when a great diversity is observed. These results are in accordance with the findings of Jiang and Tay (2010), where a higher population's diversity favored the performance and stability of the tested bioreactors for a long period of time in comparison to pure cultures, which are not robust and do not withstand the changing conditions. Eisenhauer et al. (2012) showed that microbial diversity increases the stability of microbial communities relative to both biotic and abiotic environmental parameters. So, it is important to understand how the potential relationship and interaction networks between microbial populations and physicochemical parameters are related to optimum microbial ecosystem function. Indeed, the amount of species, their diversity and distribution characteristics of the microorganisms depended on the properties of the compounds being treated (Li et al., 2013). One should take into account that NH3 is brought by the incoming air at the same time as H2S, and water from watering also bring N-NH4. Although pH values around 6–7 (conditions of the pozzolan biofilter) are regarded as optimal for gas biofiltration, it has been shown that nitrification is optimal at neutral pH (Liu et al., 2008). Unfortunately, NH3 was not measured in our study, but we are aware that it is an important factor as NH3 and H2S could be simultaneously oxidized. Nitrate addition has been used for sulfide control in WWTP for a long time. Autotrophic denitrification coupled to sulfide oxidation was regarded as one of the most important mechanism contributing to the control of sulfide contamination (Shao et al., 2010; Chang et al., 2011). Predominant species of Thiobacillus found in marble packed biofilter are affiliated among others with Thiobacillus denitrificans, a species well known for its capacity to couple the oxidation of inorganic compounds of sulfur with the denitrification. Moreover, pH variations affect microbial community composition. The present study and the work of Omri et al. (2011), show a higher bacterial diversity in lower pH values whereas a poor diversity occurred in conditions where pH values are around 5.5–6.5. Indeed, microbial catabolic activities can affect reactor pH, with either inhibitory or noninhibitory outcomes on microbial community composition. Taken together, all these preliminary results suggest that in marble biofiltration system, H2S seems to be preferentially removed by specific SOB whereas nitrifying and denitrifying bacteria such as Betaproteobacteria species (like Nitrosomonas) are less active as they are less represented. This is in adequacy with the results of Jiang and Tay (2010) who suggested that nitrifying bacteria might be affected by high loading of H2S and accumulation of the metabolic endproducts. However, their presence could be explained by the acclimatization of some nitrifying bacteria, which may sustain high loadings of H2S. Conversely, in the pozzolan biofiltration support, H2S removal is less efficient probably because of the associated limestone, which could increase the pH, leading to a decrease of SOB activity. Thus, bacteria responsible for nitrogen removal should grow preferentially on this type of material support.
Conclusion
Results of this work show that the nature of the packaging biofiltration system and environmental parameters like pH are of paramount importance for the microbial diversity and the richness of microorganisms involved in H2S removal efficiency. Moreover, SOB may comprise less known bacteria as uncultured species of prokaryotes such as Bacteroides, and Eukaryotes such as the red alga (16S rRNA plastid gene of C. caldarium) and other not-yet-cultivated organisms. High throughput sequencing technologies such as RNA sequence technology could give important metabolic information on this process.
Footnotes
Acknowledgments
The authors are grateful to the Genoscope sequencing team for the technical assistance and to ONAS Sousse-North for providing biofilter samples from the WWTP.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.