
Abstract
Abstract
This study examined the efficacy of electrokinetic remediation of chelated mine tailing soils with mixed heavy metals. Electrokinetic experiments were conducted in a nontraditional bench-reactor that contained intermediate liquid collection interfaces within treatment zones between the electrodes. Tests were conducted using 0.05 M Na2EDTA as the chelating permeant. Tap water was used in control experiments. A constant direct current voltage of 20 V (electric field≈0.625 V/cm) was applied across working electrodes for 48 h. Transient and spatial distribution of pH, conductivity, oxidation-reduction potential (Eh), and cumulative mass of the metal species in solution were measured. In all experiments, including controls, a larger portion of soluble metals were found in the anode reservoir, indicating transformation of the metals into complex species of negative charge, reverse electroosmotic advection toward anode, and/or colloidally assisted transport. Na2EDTA was expected to increase the metal extraction into the analyte as it produced negatively charged complexes with the metals. Soluble mass of the metals was markedly low in the catholyte, with heavy precipitation of metal hydroxide salts in the ensuing high pH, low Eh environment. Total removals of all three metals were either unchanged or lower, for the same duration of treatment with 0.05 M Na2EDTA than with tap water. Results showed that chelating agents, as was exemplified with a commonly used ligand in here, may not be advantageous in enhancing electrokinetic remediation of heavily contaminated mine tailings. In such substrates, metals' transport and removal regime (i.e., rate, sequence and preference of extraction) appeared to be influenced more so by the type of metal and transient distribution of the pH-Eh under the electric field than the solubilizing effect of a ligand within the treatment zone.
Introduction
A
Enhanced electrochemical processes have been shown to produce effective results in lowering the levels of contaminants in soils, sediments, and industrial waste water (Pamukcu et al., 1997; Ottosen et al., 2001, 2009; Hansen et al., 2005; Garcia-Gutierrez et al., 2007; Reddy et al., 2010). Among these, electrokinetic method is the process of applying a low-voltage direct-current electric field across the treatment zone or contaminated soil mass to transport charged particles, ionic constituents, and polar molecules (e.g., ions, water, micelles, and colloids) toward the working electrodes where they can be extracted or deposited (Acar et al., 1995; Gill et al., 2014). Other electrochemical methods take advantage of oxidation-reduction of the resident contaminants by the passage of electric current and the changing pH of the substrate. Externally triggered electrochemical processes using an applied direct current (DC) electric field to contaminated soils and sediments have been shown to aid in transformation of some metal ion species and organic molecules to their benign forms (Pamukcu et al., 2004; Alshawabkeh and Sarahney, 2005; Brosky and Pamukcu, 2013). Electrochemical remediation of contaminated soils can be enhanced further by introducing the substrate complexing agents, such as ethylenediamine tetraacetic acid (EDTA), citric acid, and S,S-ethylene-diaminedisuccinic acid (EDDS), to solubilize and make available the target substance in the soil pore space for effective transport (Yeung et al., 1996; Hosseini et al., 2011; Karim and Khan, 2012; Suzuki et al., 2014).
In this study, we evaluated the removal efficiencies of electrokinetic remediation for cadmium (Cd), lead (Pb), and zinc (Zn)-contaminated soil simultaneously, in laboratory scale with chelating agent EDTA. Electrochemical tests were carried out on a bench scale reactor to assess the effectiveness of using Na2EDTA, a strong chelating agent to enhance electrokinetic transport and extraction of mixed metals typically found at high concentrations in mine tailing soils. Disodium EDTA salt (Na2EDTA), an odorless, crystalline aminopolycarboxylic acid, is an extensively used chelator of divalent cations such as Pb2+, Zn2+, Cd2+, and Ca2+. Like many organic chelants, it behaves like a weak acid in aqueous solutions. The Na2EDTA was chosen because of its high solubility and six-fold coordination capability with metals (i.e., hexadentate ligand). For simplification EDTA is assigned the formula H4Y. When disodium salt is used, H2Y= is the complex-forming radical in the aqueous solution and reacts with cations according to the following equations (Wong et al., 1997):
As shown from reactions, EDTA creates an anionic complex (H2Y=) with metal cations. When Na2EDTA is added to the substrate, the EDTA complexing species will have the tendency to flow toward the anode electrode under the electric gradient because of its negative charge. Eligibility of electrokinetic remediation is that it may be used successfully integrated with other soil treatment technologies such as permeable reactive barrier, soil washing, and phytoremediation (Cang et al., 2011; Demir and Koleli, 2013). Additionally, its most important advantage is that it can be applied to remediation of soils contaminated with multi-metals and organic contaminants simultaneously.
Investigation
Soil sample and its characterization
The soil sample used in this study was obtained from a contaminated mine tailing site in Southern Turkey. Soil was collected from the top 30 cm layer of the area and transported to the laboratory in an airtight plastic bag. The sample was first air-dried and then crushed to pass through a 2 mm opening sieve. Standard characterization tests were conducted on the homogenized soil samples according to the ASTM procedures. Soil sample was analyzed for pH in a 1:1 (m:v) ratio of soil to water solution suspension after 1 h of contact time following ASTM D 4972-01 (2007). The electrical conductivity (EC) was analyzed in the same suspension using EC meter and oxidation-reduction potential, Eh was determined according to ASTM G200-09 (2007) developed for soils. The three major heavy metals, Cd, Pb, and Zn, targeted for remediation were analyzed following EPA 3050B method.
Electrokinetic reactor
A schematic of the electrokinetic reactor used in this study is shown in Fig. 1. The apparatus was made of an acrylic box that measured 5 cm in depth and height, and 32 cm in length. The reactor consisted of two electrode compartments, three soil chambers, and two interface liquid compartments as shown in Fig. 1. A DC power supply was used to apply constant potential of 20 V across the working electrodes. A multimeter was used to measure the transient current, and the voltage drop between the consecutive measurement locations marked as 1, 2, and 3 in the soil chambers. The interface solution sections were constructed to simulate sampling wells that allow transient measurement of metal concentrations, pH, EC, and Eh within the zone of electrokinetic mass transport.
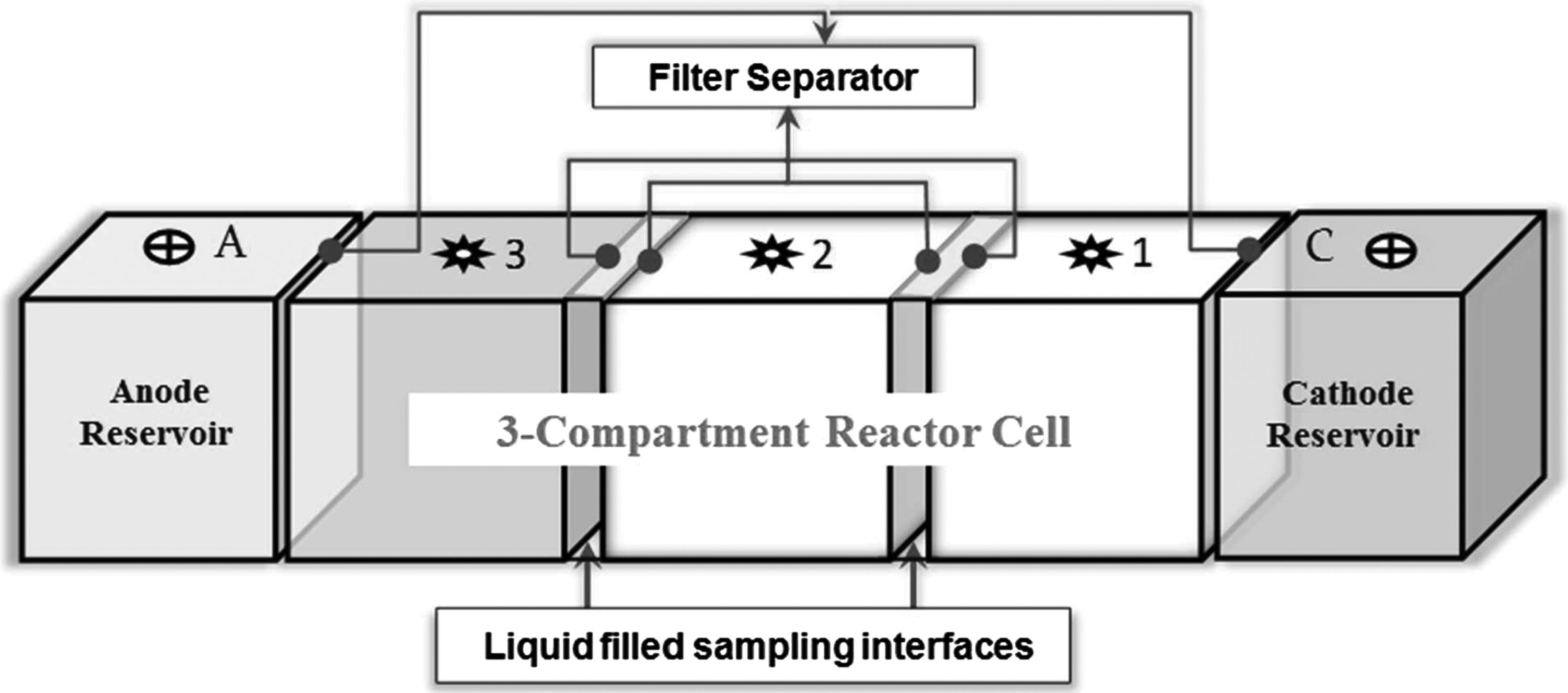
Schematic of electrokinetic experimental setup.
Air-dried soil was soaked and saturated either with tap water or 0.05 M Na2EDTA solution depending on the experiment. The wet paste was mixed by a glass rod for several minutes to achieve homogeneity.
The paste-like soil was then packed in equal mass (335 g) into each of the three soil compartments. Auxiliary titanium wire electrodes were inserted into the soil compartments to measure transient potential and current in the system. The working electrodes were platinum wire mesh plates. They were placed in the anode and cathode compartments in vertical direction facing the soil interfaces. These electrodes were 1.2×1.2 cm square and 0.08 cm thick, and connected to the DC power supply with a platinum wire. The interior liquid compartments were separated from the soil compartments by nonwoven geotextile filters. The 3 mm thick filters with a nominal permeability of 10−4 cm/s in the orthogonal direction were stretched in plexiglass frames against the soil compartments. They served to retain the soil particles while allowing the passage of water and solutes.
Experimental protocols
All experiments were carried out at room temperature and pressure for 2 days. The electrokinetic reactor was conditioned by equilibrating the tap water or the 0.05 M Na2EDTA solution in the inflow and outflow tubes connected to the electrode chambers. The solution in the tubes was kept at the same level during the experiments, thus eliminating interference of possible hydraulic gradient. A DC power supply was applied to maintain a constant 20 V (0.625 V/cm gradient across working electrodes). Accordingly, the current varied from about 120 mA to 10 mA for tap water experiment, and from 150 to 2 mA for 0.05 M Na2EDTA experiment.
During the treatment process, 10 mL samples were taken from the liquid compartments at different time periods for 48 h. The EC, pH, and redox potential (Eh), and the heavy metal concentrations of these samples were measured over the duration of the tests. All concentrations were measured by using Atomic Absorption Spectrophotometer (Perkin Elmer AAS) following the standard procedures.
Results and Discussion
Characteristic of the soil
Initial values of the physical and chemical parameters of the air-dried soils used in this study are given Table 1. Particle size distribution of soil was determined as 49%, 46.7%, and 4.3% as sand, silt, and clay, respectively. The texture classification of the soil was sandy loam (SL) according to the U.S. Department of Agriculture Classification system. Because the soil was SL, the Atterberg (consistency) limits of the soil was determined as nonplastic according to ASTM D 4318 (2000) procedure. The pH of the soil was determined to be slightly acidic at 6.29. The amount of organic matter was very low at 1–2% by mass in soil. The initial concentrations of Pb, Cd, and Zn in the soil sample were 13,365, 15,742, and 19,565 mg kg−1, respectively (Table 1). These values suggested that the mine tailing soil was highly contaminated according to the Soil Pollution Control Standards of Turkey (Soil Pollution Control Standards, 2001) with the maximum allowable limits specified as 300, 3, and 300 mg kg−1 for Pb, Cd, and Zn, respectively.
ASTM, American Society for Testing and Materials; USEPA, United States Environmental Protection Agency.
The results of the sample characterization are provided in Table 1.
Electrochemical experiments with tap water as mixing and permeating liquid
The transient distribution of the voltage gradients between working and auxiliary electrode, and the current are presented in Fig. 2.
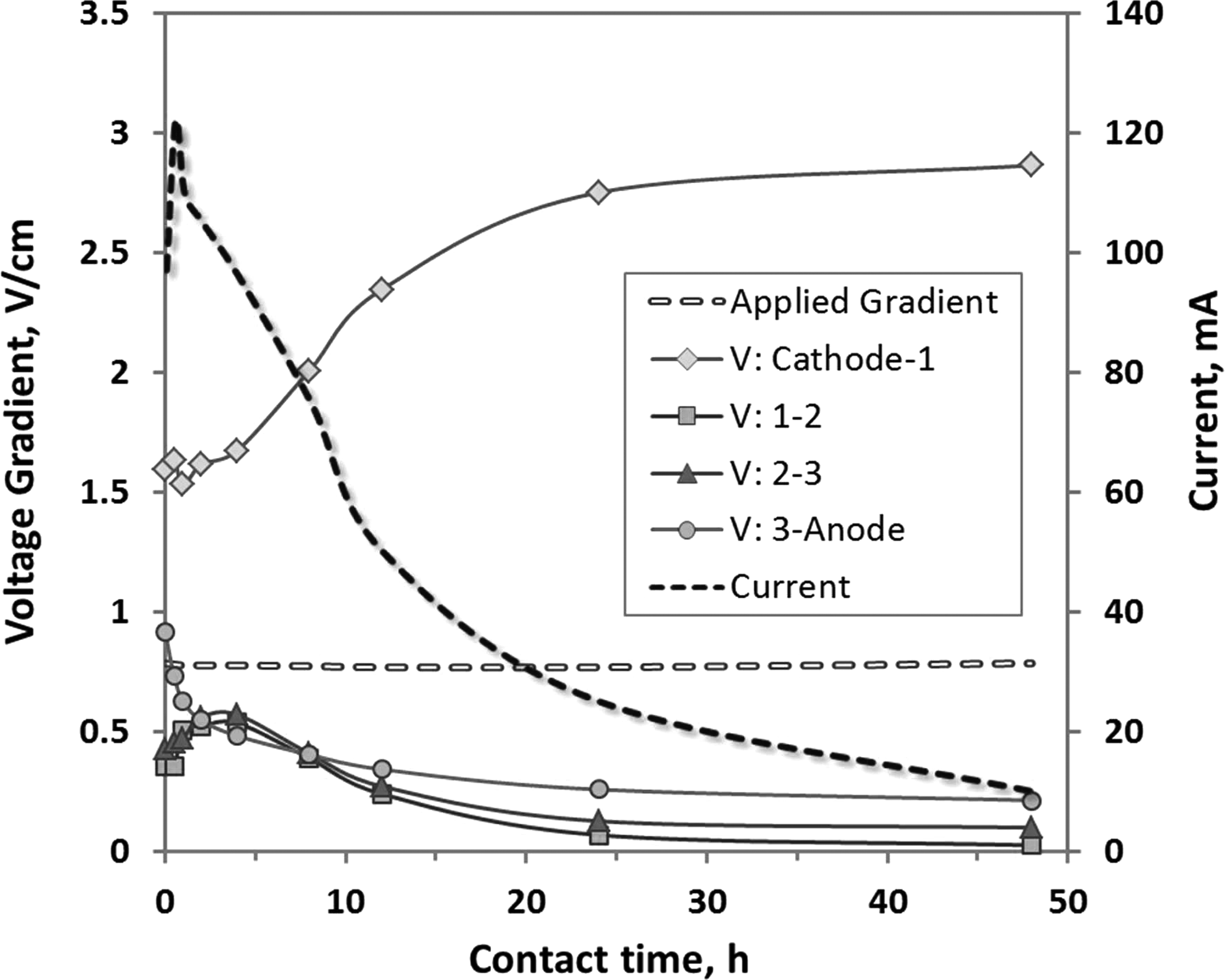
Transient and spatial distributions of voltage gradient and current in soil mixed and permeated with tap water.
The current depleted from over time, as typical of electrokinetic treatments under constant voltage. The applied potential gradient (VT) remained constant during the experiment. The potential gradients were lower at all sections except in section “Cathode-Station 1” (VC-1).
This is unlike most experimental findings using simulated media, such as kaolinite clay, where the largest potential drop occurs at the anode-soil interface of the reactor. Since kaolinite is a “cationic soil” with point of zero charge (PZC) at 2–4.6 (Miranda-Trevino and Coles, 2003; Yukselen and Kaya, 2003)—that is with affinity to retain and exchange cations—the electroosmotic flow occurs from positive to negative electrode. In cationic soils the electroosmotic flow initiates with suction generated at the liquid-soil interface at anode side and mass movement of water occur toward cathode. The voltage gradient distribution in the mine tailing soil tested in here suggests that the suction initiated flow may have started at the cathode side liquid-soil interface. The following explanation is offered for this occurrence. Although not confirmed with a mineralogical analysis, it is likely that the fines portion (clay and silt) of the mine tailing soil predominantly contain anionic minerals (i.e., gibbsite, PZC: 10)—that is with affinity to retain and exchange anions.
An anionic soil would generate flow toward anode, counteracting any transport to cathode. Under these circumstances, the mass flow of ionic constituents to anode would further be enhanced if the extracted metals are also in their anionic complex forms and migrate to the anode under the applied electric field. Although the mineral analysis of the soil sample was not conducted in this work, the flow results suggest that electroosmotic advection might have occurred toward the anode.
Figure 3 shows the transient distribution of pH, EC, and Eh in the electrode compartments and the intermedia solution locations. As expected, water electrolysis at the electrodes generates an acidic and alkaline medium respectively in the anode and cathode. The pH of solutions around the anode decreased to about 1.0 while around the cathode it increased to 11.0 by 48 h. The overall conductivity of the soil specimen was fairly low (= 0.23 mS/cm) initially, but steadily increased toward the anode (= 58 mS/cm), while it remained low at the cathode (= 1.3 mS/cm) for the duration of the test. The oxidation-reduction potential distribution appears to follow the pH distribution closely with oxidation at low pH and reduction at high pH by 48 h. It is noteworthy to observe that the entire cell exhibits a uniform positive shift in Eh for the first 2 h of treatment, while the pH distribution does not follow. This may be attributed to potential electrochemical changes occurring in the metal species or in their complex molecule forms independent of the pH during the electrokinetic treatment (i.e., losing electrons, hence turning less negative or more positive).

Transient and spatial distributions of pH, EC, and Eh in soil mixed and permeated with tap water. EC, electrical conductivity.
According to electrochemical equilibria in aqueous solutions (Pourbaix, 1974), the Pb ions would have deposited at a pH of above 9.5 as its insoluble salt, most likely as Pb(OH)2. Similarly, the Cd ions would deposit as Cd(OH)2 above pH 10, and the abundant form of zinc in the water above pH 8.5 would be Zn(OH)2. Hence, all three metals in the soil sample would have precipitated as hydroxide complexes and transformed into insoluble salts in the cathode compartment by 8 h of treatment when the pH reached above 11. The Eh value is observed to shift below the initial by 4 h of treatment and decline to −167.5 mV by 48 h in the cathode compartment. The reductive environment and the high pH point to the formation of solid products upon gain of electrons (i.e., corrosion products). Indeed substantial precipitation was observed in the cathode chamber by the completion of the 48 h of treatment.
Figure 4 shows the transient sequential distribution of the cumulative mass of three metals in solution at the electrode reservoirs and the intermediate liquid compartments. As observed, the cumulative masses of the metals in the intermediate compartment solutions (i.e., 0.36 and 0.6 ×/L from cathode side liquid-soil interface) are very similar and low in magnitude confirming that the ionic species maintain their mobility toward the electrode locations and do not concentrate or be entrapped within the system. The accumulated aqueous mass of metals in the analyte reservoir is consistently higher than that in the catholyte reservoir.
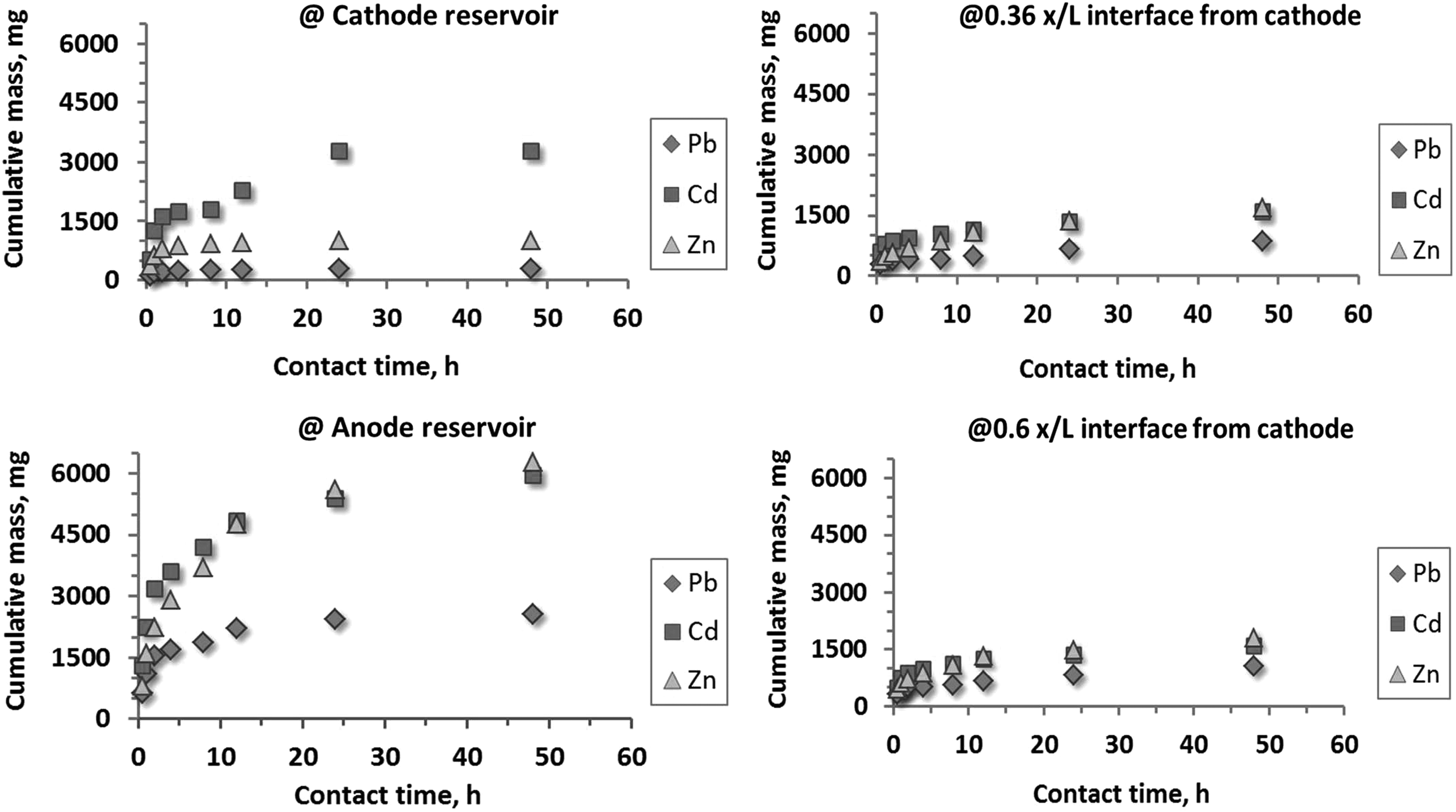
Transient distribution of cumulative mass of three metals (Pb, Cd, and Zn) in solution at four liquid sampling locations where mixing and permeating fluid is tap water.
This can be explained by combination of several potential factors: (1) the fines constituent in the mine tailing render the soil “anionic” (i.e., gibbsite) resulting in electroosmotic mass transport toward anode; (2) presence of anionic complexes of the metal ions that migrate to anode; (3) precipitation of the metal ions into insoluble salts at the supporting Eh-pH combination in the catholyte, hence their removal from solution; (4) colloidal transport of the metals into analyte. The cumulative mass of Pb in solutions is significantly lower than those of Cd and Zn. This can be explained by stronger adsorption and retention of Pb onto soil solids. Although the initial concentrations of Cd and Zn in the soil were different, the quantities of these two metals transported to the solutions are close, indicating absence of preferential extraction or competition between their transport processes.
At the completion of the 48 h treatment, the overall removal of lead, cadmium, and zinc from mine tailing soil was computed approximately as 35%, 78%, and 55% respectively.
Figure 5 shows individually the cumulative mass distribution of the three metals in solution at four sampling points along the electrokinetic reactor over the treatment duration. The mass accumulation of all the metals appears to be at highest rate for the first 2 h of treatment. Then, decreasing gradually, all mass accumulations reach a slow steady pace after 24 h. Coincidently, the Eh distribution also shows an abrupt shift to lower values after the first 2 h of treatment, indicating the effect of ensuing electrochemical changes and potential transformations of metal species on mass transport rate.

Transient distribution of cumulative mass of the individual metal ions (Pb, Cd, and Zn) in solution at four liquid sampling locations where the mixing and permeating fluid is tap water.
The mass transport relates to current and pH evolution within the system, as well. The mass of metals transported from soil to the solutions decreased with the decreasing current and with increasing pH difference across analyte and catholyte. At pH 11 and positive Eh values, the three metal ions are expected to precipitate out of solution as hydroxide salts in the catholyte. The low aqueous masses of the metals measured in the catholyte confirm that electrochemical precipitation occurs in the cathode compartment whereby the metal ion concentration in solution drops.
The migration of these metals into the anode chamber in large quantities is partially due to their solubility at low pH; possible reversed electroosmotic flow into anode chamber, and the tendency of the metal ions to form anionic complexes at the transient Eh-pH conditions during treatment. These anionic complexes would then electromigrate into the anode chamber and remain in solution at low pH.
Overall, the accumulation of Pb and Zn masses in the intermediate solutions, and the catholyte were similar. In contrast, the cumulative mass of Cd in the catholyte and analyte were markedly higher than the two internal locations. This is attributed to higher solubility of cadmium in the water than that of Pb or Zn, hence availability of more Cd for transport.
Electrochemical experiments with 0.05 M Na2EDTA as mixing and permeating liquid
In the second set of experiments, Na2EDTA was used as the mixing and permeating electrolyte instead of tap water. Figure 6 shows the transient distributions of voltage gradient and current in these tests. The current depleted to much lower values than those of the tap water case, and relatively quickly. This is plausible since Na2EDTA is expected to arrest the current carrying ions and move slower due to the larger size of the molecule.

Transient and spatial distributions of voltage gradient and current in soil mixed and permeated with 0.05 M Na2EDTA.
Voltage gradient measured in the “Cathode-Station 1” (VC-1) interval was once again significantly higher than the other sections, indicating reverse electroosmotic flow.
Figure 7 shows the transient and spatial distribution of pH, EC, and Eh at sampling locations along the reactor. The pH of the intermediate solutions decreased faster to lower values than encountered in the specimen with tap water. While the initial EC was higher in this specimen, the transient and spatial change of the index was smaller than that of the tap water case. Finally, the Eh distribution was markedly different in the Na2EDTA specimen than observed in tap water specimen.

Transient and spatial distributions of pH, EC, and Eh in soil mixed and permeated with 0.05 M Na2EDTA.
While Eh values of the intermediate solutions were lower than original in tap water specimen, these values were significantly higher than original in the Na2EDTA mixed specimens. The Eh values are highly influenced by pH, hence part of the reason for high Eh values is the low pH reached at the intermediate solutions. Furthermore, high Eh shows an oxidative environment where ionic species lose electrons to attain less negative or more positive state. Smaller negative charge would signal slower transport to anode, and larger positive charge would signal faster transport to cathode. Indeed, the transient and total mass accumulation of Cd and Zn were very close in the analyte and catholyte with Na2EDTA case, in contrast with the large separation observed in the tap water case.
Figure 8 shows the transient sequential distribution of the cumulative masses of Pb, Cd, and Zn in solution at the four liquid sampling locations. As discussed previously, the Na2EDTA reaction with the metal species produces negatively charged complexes; therefore, they are expected to be transported toward anode. In this test, the catholyte accumulation of the metals followed the same sequential order (Cd>Zn>Pb) as that of the tap water case. The accumulation in the analyte did not appear to follow this trend. With Na2EDTA, the selectivity of metal removal into analyte appears to lessen when compared to the tap water case. The mass accumulation in the intermediate solutions were similar and remained low suggesting steady transport of the metal ions away from the central region of the packed soil.

Transient distribution of cumulative mass of three metals (Pb, Cd, and Zn) in solution at four liquid sampling locations where the mixing and permeating fluid is 0.05 M Na2EDTA.
Figure 9 shows individually the cumulative mass distribution of the three metals in solution. With 0.05 M Na2EDTA mixing solution, the total removal efficiency of the metal ions were determined as 32%, 55%, and 29% for Pb, Cd, and Zn, respectively. Although slightly more Pb showed up in catholyte solution with Na2EDTA case, the total Pb removal did not change from that of tap water case in general. However, observing the rate of transport beyond the first 12 h of treatment, it appears the rate of Pb transport to anode increased with Na2EDTA. Hence, it is plausible to predict higher rate of recovery for Pb with Na2EDTA over longer duration of treatment.

Transient distribution of cumulative mass of the individual metal ions (Pb, Cd, and Zn) in solution at four liquid sampling locations where the mixing and permeating fluid is 0.05 M Na2EDTA.
The Cd mass decreased in the anolyte and showed no change in the catholyte and the intermediate collection sites when compared to the tap water case. The most appreciable change occurred with Zn, where markedly more of the Zn remained in aqueous form in the catholyte and less of it migrated into the anolyte, as shown in Fig. 9.
These results indicate that although Na2EDTA is used as the mixing and permeating electrolyte to enhance the mobilization and removal of Cd, Pb, and Zn in soil simultaneously, interactions of heavy metals and Na2EDTA may have an adverse effect on the efficiency of electrokinetic process because competitive interactions occur between divalent metal cations moved from the soil to the electrolyte. Moreover, it can be said that competition between metal cations and the presence of Na2EDTA can antagonistically affect their removal efficiencies and electrodeposition in the soil and electrolyte. Competitive interactions between metal cations and Na2EDTA (organic ligand) may either increase or decrease significantly the level of each other depending on whether the interactions are synergistic or antagonistic. Synergistic effect implies that increasing the level of one of the interacting element increases the level of the other, while antagonism implies the converse (Wuana et al., 2014). The results of this study have demonstrated that competition between heavy metals and organic ligand has led to decrease of metal removal efficiency. There also exists evidence of an antagonistic interaction between Zn and Cd (Oliver et al., 1999). Cadmium and Zn appear to compete for certain organic ligands in vivo, Cd competes with Zn in forming protein complexes through antagonistic association between the two metals (Thavamani et al., 2011; Wuana et al., 2014).
In both cases of mixing fluids (i.e., tap water and 0.05 M Na2EDTA), the mass transport of metals appears to reach a steady state rate after 24 h of treatment at all sampling locations. The fast transport into the electrode reservoirs and the interface chambers cease within the first 2 h of treatment in all tests regardless the pore fluid chemistry. This suggests that the available soluble portion of the metals in the pore space remains unaffected by the pore fluid chemistry during fast transport. The smaller mass accumulation in the anode chamber with Na2EDTA in the pore fluid is potentially due to partial arrest of the metal in the pore fluid by the EDTA molecules resulting in slower transport. In contrast, the steady state rate of metal accumulation beyond the fast rate duration (i.e., after 24 h) varies with metal type and the pore fluid chemistry. While Cd and Zn transport appears to be less affected, the Pb transport rate to anode significantly increases with Na2EDTA. Hence, it is plausible to predict higher rate of recovery for Pb with EDTA over longer duration of treatment.
In both cases the metals extraction into catholyte followed the order of Cd>Zn>Pb. A similar trend prevailed in the intermediate solution locations with Cd showing up in highest quantity consistently. In contrast, metal extraction into anode appeared to be influenced by the chemistry of pore fluid, as oxidized or the chelated species of the metals tended to accumulate at the anode at different rates of transport. Finally, observations of precipitated mass in the cathode chamber confirmed the arriving metals to collapse into metal hydroxide salts, hence removed from solution. Due to extreme high precipitation, it is highly likely that the permeability of the nonwoven geosynthetic filter would have been compromised at later stages of the treatment and result in slowing down the transition of cations into catholyte.
Conclusions
Transient and spatial distribution of electrochemical indices (Potential gradient, current, and pH, EC, and Eh) and cumulative masses of three target heavy metals (Pb, Cd, and Zn) were evaluated for field retrieved mine tailing soil in electrochemical treatment tests.
Two tests were conducted using different mixing/permeant fluids—tap water and 0.05 M Na2EDTA—for comparative evaluation of removal efficiencies and identification of important electrochemical indices. In both cases of mixing fluids the mass transport of metals appeared to reach a steady state rate after 24 h of treatment at all sampling locations. The fast transport into the electrode reservoirs and the interface chambers ceased within the first 2 h of treatment in all tests regardless of the pore fluid chemistry. This duration of treatment (2–4 h) also coincided with marked changes in the Eh and pH distributions observed within both specimens, rendering these indices important markers of transport behavior. The available soluble portion of the metals in the pore space remained unaffected by the pore fluid chemistry during fast transport. The metal transport and accumulation beyond the fast rate duration (i.e., after 2–4 h) varied with metal type and the pore fluid chemistry. In both cases the metals extraction into catholyte followed the order of Cd>Zn>Pb. In contrast, metal extraction into anode appeared to be influenced by the chemistry of pore fluid, as oxidized or the chelated species of the metals tended to accumulate at the anode at different rates of transport. The lower mass accumulation in the anode chamber with Na2EDTA in the pore fluid was attributed to slower transport due to the larger size of the resulting complex molecules. Finally, observations of precipitated mass in the cathode chamber confirmed the arriving metals to collapse into metal hydroxide salts in the ensuing high pH-low Eh environment.
It is concluded that complex interactions among the metals and organic ligand can antagonistically affect electrokinetic remediaton in the soil and electrolyte because of their lower removal efficiency.
Footnotes
Acknowledgments
This work was funded by The Scientific and Technological Research Council of Turkey (TUBITAK-BIDEB 2214). The Department of Civil and Environmental Engineering at Lehigh University is acknowledged, where all the equipment development, testing, and analysis for this work were funded. Dan Zeroka of Lehigh University is kindly acknowledged for the modification of the electrokinetic reactor apparatus.
Author Disclosure Statement
No competing financial interests exist.