
Abstract
Abstract
Hydrogen peroxide (HP) and its derivatives can serve as useful bulk oxidants for soil and water treatment provided activation is supplied. Solutions of a model compound methylene blue (MB) were treated with HP, urea hydrogen peroxide (UHP), sodium perborate (SPB), sodium percarbonate (SPC), or calcium peroxide (CP). In unbuffered systems, addition of HP or UHP was ineffective in decolorizing MB, whereas addition of SPB or SPC was moderately effective due to the known ability of borate or carbonate ions, respectively, to activate H2O2. Addition of CP caused moderate decolorization as well, but base hydrolysis contributed due to the high pH (∼12). Decolorization of MB in HP, UHP, SPB, and SPC systems was greatly accelerated when mixtures were buffered at pH 10 with phosphate (25 mM) compared to mixtures where pH was unadjusted, NaOH adjusted, or carbonate adjusted. Decolorization correlated strongly with decomposition of active peroxide (AP), implying a shared pathway. Decolorization was highly inefficient relative to AP decomposition, however. This study represents the first example, to our knowledge, demonstrating activation of H2O2 by phosphate toward oxidation of organic compounds. It is suggested that the reactive oxidant species responsible for decolorization and AP loss in the presence of phosphate is, or is derived from, HPO52−.
Introduction
H
Some solid peroxides have “built-in” H2O2 activators (see following paragraph) and also provide an alkaline environment conducive to certain oxidation pathways. Solid peroxide reagents have been used as oxidants in organic synthesis and a source of molecular oxygen for aerobic bioremediation in the subsurface, but few reports exist on their use for water or soil treatment (Waite et al., 1999; Arienzo, 2000; Kao et al., 2003; Pirnie et al., 2006; Cravotto et al., 2007; Northup and Cassidy, 2008). One in particular found that CP and magnesium peroxide could degrade polychlorinated biphenyl-containing oil (Aroclor 1016) in soil (Goi et al., 2011). The reagents SPC, SPB, and CP are relatively inexpensive, nontoxic, stable, and safe to handle and store.
While HP can react at high pH slowly with some compounds, notably dyes (Thompson et al., 1993a, 1993b; Zeronian and Inglesby, 1995; Spiro and Griffith, 1997; Brooks and Moore, 2000; Katafias et al., 2010), it is inherently a weak oxidant and thus requires activation. Activators can be selected to initiate one-electron (free radical) or two-electron (electrophilic/nucleophilic) pathways of oxidation. The radical pathway is initiated by photolysis (Li et al., 2007; Yang et al., 2014) or reaction with a low-valent transition metal, usually iron (Watts et al., 1990; Pignatello and Baehr, 1994; Anipsitakis and Dionysiou, 2004; Pignatello et al., 2006), and generates, most importantly, the hydroxyl radical (•OH) as the reactive oxidant species (ROS). A radical pathway is also involved in the conversion of the peroxo unit in peroxymonosulfate (HSO5−) or peroxydisulfate (S2O82−) derivatives of H2O2 to •OH and/or the sulfate radical (SO4•−) by heat, light, or low-valent transition metals (Tsitonaki et al., 2010; Zhang et al., 2015). Hydroxyl and SO4•− ROS react with organic compounds with little selectivity.
The two-electron pathway of oxidation is initiated by carbonate (Richardson et al., 2000; Yao and Richardson, 2000) or borate (McKillop and Sanderson, 1995; Gomez et al., 2007; Lobachev et al., 2010), which coordinates the peroxo unit. The peroxo unit in carbonate or borate complexes behaves as an electrophilic or nucleophilic ROS, respectively. Carbonate or borate activation has been used to oxidize a variety of compounds, including alkenes (Yao and Richardson, 2000), methionine (Richardson et al., 2003), aryl sulfides (Richardson et al., 2000; Yao and Richardson, 2003; Gomez et al., 2007), diethyl sulfide (Lobachev et al., 2010), methyl phenyl sulfide (Fakhraian and Valizadeh, 2010), methylphosphonothioate (Wagner and Yang, 2002), and methyl blue (Xu et al., 2011).
The model compound employed in this study is methylene blue (MB), which is used commercially for dying cotton, wool, and silk (Rafatullah et al., 2010). Removal of organic dyes from waste water is necessary for reasons of aesthetics and toxicity. Solid peroxides CP, SPB, SPC, UHP, or HP were added to MB solution with or without buffer, and the rates of MB decomposition were measured. In this study, we report the novel observation that phosphate ions greatly accelerate MB decomposition by these reagents and HP itself under alkaline conditions.
Materials and Methods
Materials
MB, CP (75% CaO2), SPC (available H2O2, 20–30%), SPB monohydrate, and UHP (15–17% active oxygen content) were purchased from Sigma-Aldrich. HP reagent (30% by weight), trisodium phosphate dodecahydrate (≥98%), sodium bicarbonate, sodium hydroxide, and other chemicals were of reagent grade. All solutions were prepared in water from an ultrapure system (Barnstead) (resistivity >18.2 MΩ·cm).
The peroxide content of solid peroxides was determined by titration in acidic solution (6% HCl and 14% H3PO4) by 0.5 N KMnO4 (Schumb et al., 1955). The determined active peroxide (AP) contents of CP, SPB, SPC, and UHP solids were 10.79 ± 0.08, 9.53 ± 0.06, 8.41 ± 0.09, and 10.39 ± 0.02 mmol H2O2/g, respectively—in all cases within the range reported on the product label.
Experimental procedures
In a typical experiment, 20 mL of water containing the desired amounts of MB and buffer were placed in a 40-mL amber glass vial with a Teflon-lined silicone septum screw cap at room temperature (20 ± 2°C). The initial MB concentration was 10 mg/L. The pH 10 buffer was 25 mM sodium carbonate or 25 mM sodium phosphate. The pH 7 buffer was 25 mM sodium phosphate. The reaction was initiated by adding 40 mg of the reagent to the vial at a concentration of 2.0 g/L. The maximum theoretical AP concentration in solution was thus ∼20 mM. Experiments were done in duplicate.
Degradation of MB was monitored by periodically withdrawing 2 mL of the aqueous phase and immediately recording the absorption spectrum using a Hewlett-Packard 8452A spectrophotometer. In the case of CP, the sample had to be prefiltered through a 0.2-μm syringe filter. Thus, multiple vials were set up and duplicate vials were sacrificed for analysis at each time point. The concentration of MB was calculated by comparing its maximum absorbance at 656 nm with calibration standards. Solution-phase AP concentration was monitored by diluting a separate 2-mL sample of the supernatant (filtrate for CP) into 25 mL of 6 mM ammonium metavanadate and 75 mM H2SO4, which forms a red-orange peroxovanadium cation within 20 min with maximum absorbance at 450 nm (Nogueira et al., 2005). The absorbance at 450 nm was corrected for MB absorbance and compared with H2O2 calibration standards. MB absorbance at 450 nm was calculated from the known concentration of MB determined by its absorbance at 656 nm and the MB molar extinction coefficient at 450 nm assessed from calibration standards.
Results and Discussion
Background on speciation and behavior of the peroxide reagents in water
The compound known as SPB is the sparingly soluble disodium salt of the heterocycle, 1,4-diboratetroxane, B2O4(OH)42− (McKillop and Sanderson, 1995). In aqueous media, SPB hydrolyzes rapidly through various peroxoborate intermediates to give a moderately alkaline solution (pH ∼10) containing H2O2, B(OH)3, and B(OH)4− in equilibrium with a small concentration of monoperoxyborate HOOB(OH)3−. The peroxyborate species is thought to act as a nucleophilic oxidant (McKillop and Sanderson, 1995; Lobachev et al., 2010). The material known as SPC is the peroxyhydrate of sodium carbonate, best written as 2Na2CO3·3H2O2 (McKillop and Sanderson, 1995). It is moderately soluble in water (150 g/L) and liberates H2O2, HCO3−, and CO32− in equilibrium with a small concentration of peroxycarbonate HOOCO2− (Richardson et al., 2000). The distal oxygen of HOOCO2− seems to serve as an electrophilic center (Richardson et al., 2000). Bicarbonate activation has been used to oxidize dyes (Xu et al., 2011) and organosulfur compounds (Richardson et al., 2000, 2003; Yao and Richardson, 2003).
UHP complex (a.k.a., carbamide peroxide) is a perhydrate of urea that contains about 36% H2O2 by mass present as guest molecules densely packed in parallel pores of the urea host structure (Harris, 1997). It is slightly soluble in water (0.05 g/mL), liberating H2O2 as it dissolves. CP (“CaO2”) is a mixture of Ca(OH)2 and Ca(O-O) that is used commercially as a bleaching agent, disinfectant, and controlled source of dioxygen in agriculture, aquaculture, and aquifer restoration (Waite et al., 1999; Kao et al., 2003). It is only sparingly soluble in water. Aqueous suspensions of CaO2 become highly alkaline (pH 12–13) and slowly decompose to O2. At lower pH, CaO2 reacts with water to release H2O2 and Ca(OH)2(s) (Northup and Cassidy, 2008).
MB decolorization by peroxides without pH adjustment
Addition of HP or UHP to MB solutions had little effect on pH (initial pH, 5.66; final pH, ∼6.0). Addition of the other peroxides raised the pH to 9.70–10.02 (SPB), 9.94–10.38 (SPC), or 12.06–12.34 (CP). The rise in pH took <1 h; the pH was then stable over the remainder of the 29-h reaction period.
Figure 1 shows the change in MB and AP concentrations after adding peroxides to solutions of unadjusted pH over the 29-h reaction period. For UHP and HP, the MB and AP concentrations declined by <5%, consistent with the weak reactivity of H2O2 toward MB under acidic or neutral conditions in the absence of activators (Katafias et al., 2010; Xu et al., 2011).

MB decolorization and AP decomposition after addition of various peroxides, HP, UHP, SPB, SPC, or CP. Without pH adjustment (
The declines of MB and AP were much greater for SPB, SPC, and CP compared to UHP and HP (Fig. 1). For SPB, MB declined by 28% and AP by 26.5%. For SPC, MB declined by 52% and AP by 79%. In each case, the absorbance bands of MB in the visible region of the spectrum decreased progressively with time and no new bands appeared, consistent with oxidative bleaching (Fig. 2). The ROS could be perhydroxide, peroxoborate, or peroxycarbonate ions, as the case may be (McKillop and Sanderson, 1995; Katafias et al., 2010; Lobachev et al., 2010). For CP, MB decreased by 49%, whereas AP was found at a low concentration (0 ∼ 0.3 mM) throughout the 29-h period since this reagent continuously releases H2O2 (Northup and Cassidy, 2008). Because CP raised the pH two units higher than the other reagents, it is possible that base (OH−)-catalyzed hydrolysis contributed to MB decolorization. This possibility is borne out by the spectra in Fig. 2, which show a new absorbance band around 500–600 nm, similar to the band reported when MB was transformed in strong base (0.6 M OH−) in the absence of oxidants (Katafias et al., 2010), and is indicative of the formation of new dyes, such as azure B, azure A, azure C, and methylene violet.

Spectral changes of MB solution after adding
MB decolorization by HP in buffered solutions
Figure 3 presents the results of experiments designed originally to determine whether base-catalyzed hydrolysis contributes to MB loss and whether added carbonate could activate H2O2. The dye is stable in the absence of HP both at pH 7 in phosphate buffer and at pH 10.0 in phosphate or carbonate buffer, indicating stability to hydrolysis. The MB concentration decreased by ∼16.5% in HP at pH 10 adjusted with NaOH, indicating some reactivity toward perhydroxide (HO2−) (Katafias et al., 2010). The MB concentration decreased by 34.4% in HP buffered at pH 10 with carbonate, indicating activation by carbonate to form the ROS, HCO4−, and/or CO42−. Assuming the pKa of HCO4− is the same as bicarbonate HCO3− (10.32), the molar ratio of HCO4− and CO42− is estimated to be ∼2:1 at pH 10.
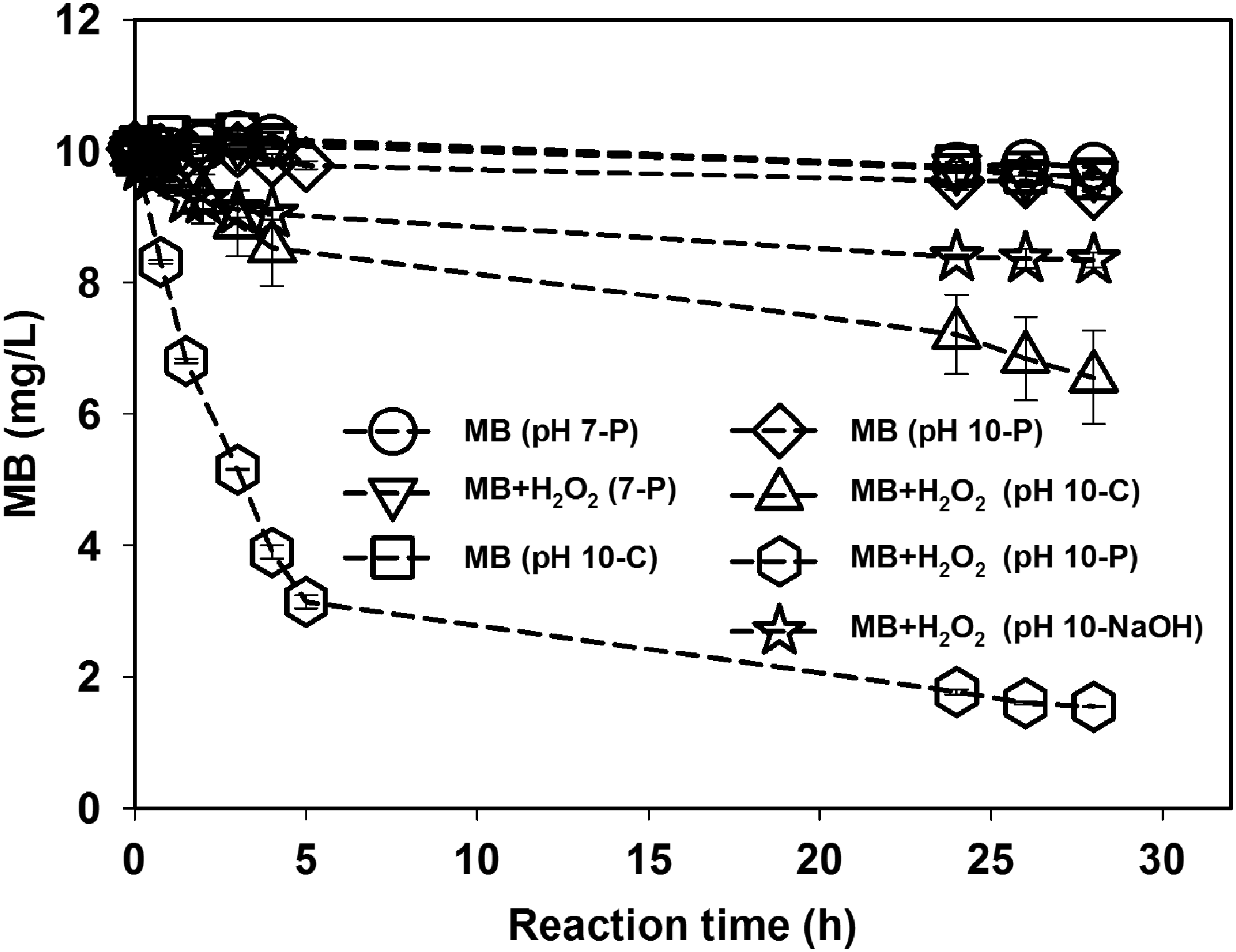
MB decolorization by HP (H2O2) at pH 10 buffered by carbonate (pH 10-C) or phosphate (pH 10-P), or adjusted by sodium hydroxide (pH 10-NaOH), and the corresponding control treatments at pH 7 buffered by phosphate (pH 7-P). Initial [MB] = 10 mg/L, [H2O2] = 20 mM, buffer concentration = 25 mM. Lines are to guide the eye. Error bars represent the standard deviation.
More surprisingly, MB concentration decreased by 84.5% in HP buffered at pH 10 with phosphate. Thus, the results of Fig. 3 show clearly that some species present in phosphate buffer at pH 10 is (are) more reactive than HO2− and HCO4−/CO42−. Catalysis by impurities of transition metal ions such as iron can be ruled out due to the absence of reactivity of HP at pH 7 in the absence (Fig. 1) or presence (Fig. 3) of phosphate.
MB decolorization by peroxides in alkaline phosphate solutions
Figure 1c and 1d show the results of MB oxidation by the various peroxide reagents added at the rate of 2.0 g/L in 25 mM phosphate solution. The pH was 9.86–10.00 (HP), 9.99–10.03 (UHP), 10.01–10.31 (SPB), 10.40–10.54 (SPC), and 12.19–12.25 (CP). First, comparing Fig. 1c with 1a, the results show that phosphate strongly accelerates MB oxidation over the first 5 h by all peroxide reagents, except for CP. The reagents HP and UHP are especially affected. Phosphate increased the final extent of decolorization markedly for HP and UHP (to 78.0% and 76.9%, respectively), slightly for SPB (by 24.0%), and not at all for CP and SPC. Second, comparing Fig. 1d with 1b, the results show qualitatively that the rate of AP decomposition is greatly accelerated by phosphate, except for CP, which generates a steady-state source of H2O2. The leveling-off phase of MB decolorization appears to commence with the exhaustion of AP after a few hours.
Rate constants and stoichiometry for AP decomposition and MB decolorization
Figure 4 shows that MB decolorization strongly correlates with AP consumption (R2 > 0.927, p < 0.01) for HP, UHP, SPB, and SPC treatments, suggesting that AP decomposition and MB decolorization are linked through a common intermediate. The slope represents the moles of AP consumed per mole of MB decolorized, a pseudo stoichiometry. In phosphate-buffered media at pH ∼10, the pseudo stoichiometry of AP:MB follows the order, SPC (1,420 ± 90) > SPB (1,037 ± 22) > HP (850 ± 35) > UHP (788 ± 39). In unbuffered media, the slope for SPC (771.7 ± 30.7) and SPB (387.7 ± 36.3) is smaller than with phosphate buffer. Thus, phosphate accelerates MB oxidation, but at the expense of greater AP decomposition.
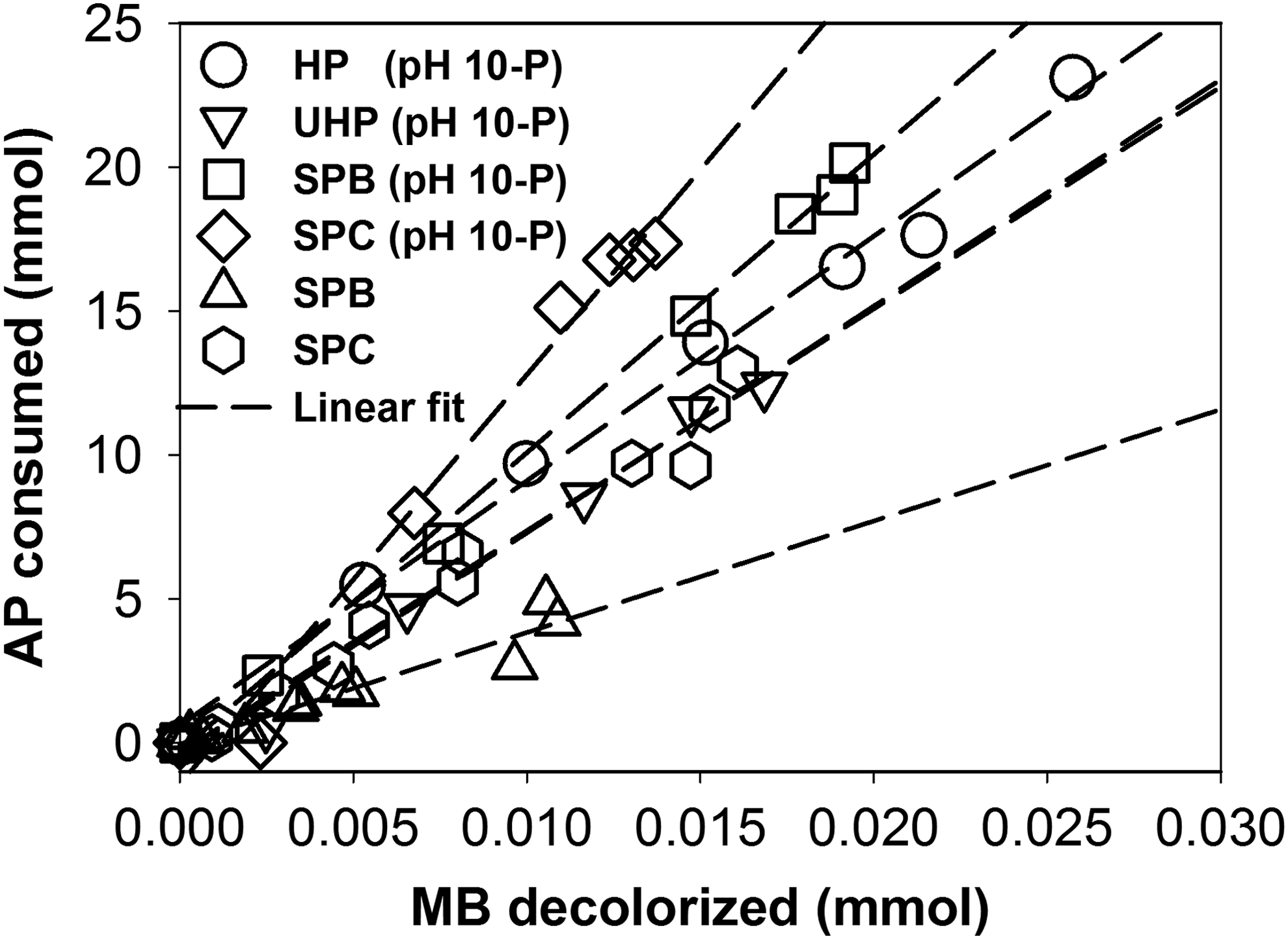
Correlations between MB decolorized and AP decomposed after addition of HP, UHP, SPB, SPC or CP. Data for all sampling times are included in each case. Data labeled pH 10-P were from experiments at pH ∼10 in 25 mM phosphate buffer. Lines represent linear least-squares fit.
Observed first-order rate constants for MB degradation (kMB) and AP decomposition (kAP) were obtained by fitting data in the initial 4–5 h reaction period to a first-order decay rate law and they appear in Table 1. Both kAP and kMB are much smaller in unbuffered than phosphate-buffered solution for all reagents except CP. The kMB in unbuffered solution at pH ∼ 10 follows the order, SPC > SPB >> HP > UHP, with a spread of 41 (0.036 × 10−5–1.49 × 10−5/s). This order strongly suggests that carbonate, and to a lesser extent borate, serve to activate H2O2. By contrast, the kMB in phosphate-buffered solution at pH ∼10 follows the order, HP > SPB > UHP > SPC, with a spread of only 2 (3.18 × 10−5–6.37 × 10−5/s). The order and low spread suggest that phosphate activates H2O2 originating from the dissolution/hydrolysis of the peroxide reagent and dominates over any activation by carbonate or borate. The kMB values for CP are similar with or without phosphate, consistent with the principal mechanism being alkaline hydrolysis, as discussed in MB Decolorization by Peroxides Without pH Adjustment section.
-P represents with phosphate buffer.
pH is the average value during the experiment.
kAP and kMB are the observed first-order rate constants for AP decomposition and MB degradation, respectively.
R2 is the coefficient of determination for rate law fitting plot. Dash (—) means the first-order rate law is poorly fit.
AP, active peroxide; CP, calcium peroxide; HP, hydrogen peroxide; MB, methylene blue; SD, standard deviation; SPB, sodium perborate; SPC, sodium percarbonate; UHP, urea hydrogen peroxide.
The kAP in phosphate-buffered solution at pH ∼10 follows the order, SPC > SPB > HP > UHP, with a spread of 5.3 (5.17 × 10−5–27.5 × 10−5/s). This supports the participation of carbonate and borate ions in the decomposition of H2O2. For SPB and SPC treatments, while kAP values are 38 and 12 times greater, respectively, in unbuffered solution than in phosphate-buffered solution, kMB values are only about 6.5 and 2.1 times greater, respectively, in phosphate-buffered than in unbuffered solution. This again underscores that phosphate increases MB decolorization at the expense of even greater AP decomposition. Both kAP and kMB are reduced for UHP relative to HP. This implies that urea plays some role in the generation of the ROS.
Mechanistic considerations
Borate and carbonate ions are clearly activating H2O2/HO2− in the SPB and SPC systems, respectively. However, phosphate is activating the UHP and HP systems and providing supplemental activation in the SPB and SPC systems, as well. This study represents the first example, to our knowledge, demonstrating significant activation of H2O2 toward oxidation of organic compounds by alkaline phosphate. In fact, past reports teach away from any activation by phosphate because they show no phosphate-induced acceleration of H2O2 oxidation of organic compounds at pH 7. Fakhraian and Valizadeh (2010) found that certain oxo anions increased the pseudo first-order rate constant for HP oxidation of methyl phenyl sulfide to the sulfoxide (k1) and the sulfone (k2) in 50:50 ethanol-water containing 4% Triton X-405 surfactant. The order in k1 was HCO3− > HSO4− > urea > H2PO4−; and the order in k2 was HCO3− > HSO4− > H2PO4− > urea. Unfortunately, rate constants for the corresponding unactivated case were not reported, so it is not clear whether activation by phosphate, however slight, was actually taking place. Sanchez et al. (2001) studied the role of phosphate in HP oxidation of 2,4,6-trichlorophenol (TCP) and other organic compounds catalyzed by iron(III) tetrasulfophthalocyanine (FePcS) at pH 7. They proposed the intermediacy of a peroxyphosphate—FePcS complex on the basis that no reaction occurred without FePcS, and that peroxymonophosphate prepared from peroxydiphosphate hydrolysis in nitric acid was inactive without FePcS. Lou et al. (2014) found that addition of phosphate solution (pH 7) “activated” peroxymonosulfate (as the commercial product, Oxone®, KHSO5·0.5KHSO4·0.5K2SO4) toward oxidation of acid orange 7, rhodamine B, and TCP; however, the observed effect may have just been due to acid neutralization by addition of the phosphate buffer since the commercial reagent is highly acidic and the rate increased steeply with increasing pH. Nevertheless, phosphate had no activating effect on HP at pH 7.
Whatever species is responsible for MB oxidation in alkaline phosphate media may also be responsible for AP decomposition. A possible candidate is the peroxo species HPO52−. Reaction between HPO52− and MB may be through O atom transfer resulting in the oxidation of MB, similar to the reaction of peroxymonocarbonate with alkenes and cysteine (Yao and Richardson, 2000; Regino and Richardson, 2007). Monoperoxyphosphoric acid [pKa ∼1.1, ∼5.5, ∼12.8 (Battaglia and Edwards, 1965)] made from addition of H2O2 to P2O5 is known to oxidize some organic compounds under acidic conditions (Ogata et al., 1979, 1982; Zhu et al., 2003), but neither rates nor mechanism of AP decomposition resulting from this species are known. The decomposition of H2O2 is accelerated by carbonate ion and with increasing pH, presumably by a peroxycarbonate intermediate (Lee et al., 2000), but the mechanism of decomposition is unknown.
Conclusions
We have shown efficient decolorization of MB solution by applying solid peroxides or HP under moderately alkaline condition without metal catalysts and in the presence of phosphate. Alkaline conditions proved to be essential. While borate and carbonate activate H2O2/HO2− in alkaline systems, phosphate appears to be superior. The strong correlation between MB decolorization and AP decomposition implies a shared pathway. Phosphate significantly increases MB decolorization, but at the expense of even greater AP decomposition. A peroxyphosphate species such as HPO52−, or a derivative of the same, is likely responsible for both MB oxidation and AP decomposition. This study is the first, to our knowledge, to demonstrate activation of H2O2 toward oxidation of organic compounds by alkaline phosphate. The mechanism and possible practical applications of peroxides activation by phosphate warrant further investigation.
Footnotes
Acknowledgments
This work was funded, in part, by the U.S. Department of Agricultural, National Institute of Food and Agriculture through the Hatch program. We are thankful for the financial support from the Chinese Scholar Council for the visiting scholarship of B.Y. to the Connecticut Agricultural Experiment Station and the University of Massachusetts.
Author Disclosure Statement
No competing financial interests exist.