
Abstract
The aim of this article is to study the sodalite synthesis from sepiolite through an alkali fusion method followed by hydrothermal process, and to investigate its application in heavy metal removal. The fused precursors were prepared through mixing sepiolite with sodium hydroxide (NaOH) and potassium hydroxide (KOH) activators, at 650°C. Hydrothermal reactions were performed at 100°C, 140°C, 180°C, and 220°C. Under the hydrothermal treatment at 140°C, pure sodalite 1 was formed from fused precursor of sepiolite-NaOH, for 48 h, while pure sodalite 2 was synthesized from fused mixture of sepiolite and KOH, at 180°C for 72 h. Pure sodalites and raw material were characterized by x-ray diffraction (XRD), scanning electron microscope (SEM), and Fourier transform infrared spectroscopy (FTIR) analyses. The potential of two sodalites for removal of Fe3+, Cr3+, and Cd2+ cations from 0.001, 0.01, and 0.1 M aqueous solutions was evaluated through a series of batch experiments. The optimum adsorption of Fe3+ and Cr3+ was achieved from 0.001 to 0.01 M solutions. In contrast, Cd2+ was removed most efficiently from 0.1 M solution. In terms of contact time, the maximum adsorption amount from 0.001 M solutions was achieved between 1 and 2 h for pure sodalite 1 and between 30 min and 1 h for pure sodalite 2. The highest adsorption rate from 0.01 to 0.1 M solutions was observed between 30 min and 1 h, for both pure sodalite 1 and pure sodalite 2. Sepiolite was shown to be successfully used as raw material for formation of pure sodalite, and subsequently, pure sodalite has considerable capability to be used for environmental cleanups.
Introduction
Sodalite, Na8
Previous Investigations on Zeolite Synthesis
A-Kln, activated kaolinite; Bnt, bentonite; Ccn, cancrinite; C-Kln, calcined kaolinite; E-Prt, expanded perlite; FA, fly ash; H-Kln, heated kaolinite; Hsdl, hydrosodalite; K-Sdl, K-sodalite; KOH, potassium hydroxide; NaOH, sodium hydroxide; Na-Sdl, Na-sodalite; Opl, opal; Sdl, sodalite; T-Kln, treated kaolinite; Zeo 4A, zeolite 4A; Zeo LTA, zeolite A, linde type A; Zeo N, zeolite N; Zeo Na-P1, zeolite Na-P1; Zeo Na-P1, zeolite Na-P1; Zeo Na-X, zeolite Na-X; Zeo X, zeolite X.
Clay minerals can be considered natural sources of alumina-silica for synthesis of zeolites due to their low price and availability. Sepiolite clay with an ideal structural formula of Mg8Si12O30(OH)4(OH2)4.8H2O is a trioctahedral phyllosilicate whose octahedral sites have been occupied by Mg (Brauner and Presinger, 1956; Inagaki et al., 1990). This clay mineral is considered an end member of palygorskite-sepiolite group of minerals with layer-ribbon structures. These minerals can also be regarded as intermediates between pyroxenes and amphiboles with the chain/ribbon structure, and smectite with structure (Velde, 1985; Jones and Galán, 1988).
The stability of the palygorskite-sepiolite mineral group has been studied under laboratory conditions showing that these fibrous minerals directly transform to other clay minerals. On this subject, Golden et al. (1985) reported that palygorskite treated with sodium hydroxide (NaOH) solution (1 mmol) can be transformed into smectite through hydrothermal reactions at 150°C, for 24 h.
Hazardous chemical substances such as heavy metals or organic compounds give rise to pollution in the environment and drastic deterioration of ecosystems. Among various types of pollution, the contamination of heavy metals causes serious health problems, since they are accumulated in living tissues and affect food chain (Volesky, 1990). In light of this, iron causes the widest damage to the environment, because excessive amount of Fe3+ in public water supplies gives rise to turbidity, and an unpleasant taste and odor. The supporting role of this metal for the growth of iron bacteria brings about clogging the pipes and increasing the flow characteristics (Ostroski et al., 2007). Chromium is one of the toxic heavy metals whose trivalent form, Cr3+, is chiefly found in natural environments. This metal's compounds are largely bound to the floating particles in water and have a monumental influence on drinking water quality. In terms of toxicity, chromium (III) is retained to a greater extent in lungs than is chromium (VI) (Edel and Sabbioni, 1985) and its compounds were identified to increase the risk of bronchiogenic cancer (Tandon, 1982). Cadmium is one of the most toxic metals even in low concentration. It is frequently formed in waste streams discharging from metallurgical alloying, ceramics, and sewage sludge. The toxicity of cadmium causes several disorders such as lung cancer, anemia, skin, pulmonary edema, brain damage, and bone and heart diseases (Voegelin and Kretzschmar, 2003; Deng and Ting, 2005; Mohan et al., 2005; Tsang and Lo, 2006).
Over the years, techniques such as chemical precipitation, filtration, solvent extraction, reverse osmosis, and chemical oxidation or reduction have been used to clean up wastewaters (Volesky, 2001). However, ion exchange is recently investigated as a beneficial and economic process for treatment of metal-bearing effluents.
Among the materials that have been applied for pollutant removal from water and wastewater, zeolites play a momentous role. They have three-dimensional open microporous structures consisting of aluminum, silicon, and oxygen with regular arrangement of SiO4− and AlO4− tetrahedral units. Their tetrahedral units construct tunnels, channels, and cavities whose intersections lead to formation of regular pores (Zaarour et al., 2014; Shirani Lapari et al., 2015). This regular arrangement causes them to have negative charge balanced with cations namely Na+, K+, Ca2+, or Mg2+. Such properties effectuate their numerous applications like acting as molecular sieves, using as catalyst (Pal and Bhaumik, 2013), and having high adsorption capacity (Stein et al., 1992).
Synthesis of zeolites is a promising field of interest since synthetic zeolites have unique properties well adjusted to the environmental and industrial applications. Consequently, they have considerable capacity to remove various contaminants with wide range of molecular sizes from wastewaters (Wang et al., 2012; Li et al., 2015).
This study has been introduced in conformity with the following objectives: (1) evaluation of sepiolite potential to be used as a raw material for synthesis of pure zeolites that could be considered in industrial and environmental applications, (2) application of alkaline fusion approach as a tentative technique for the formation of pure zeolites, (3) investigation of duration and activators in sense of their impacts on the synthesis process through usage of various types of activator and establishing diverse periods of contact time, and (4) detection of the pure zeolite capacity for heavy metal removal from wastewater through establishing the batch-type experiments, taking the advantages of synthetic monometallic solutions.
Materials and Methods
Materials
Sepiolite, which was sampled from Eeliato Sepiolite Mine, a Pliocene–Quaternary sepiolite deposit located at Fariman County, NE Iran, was used as raw material.
NaOH, as pellets (97.5%, BDH Laboratory Supplies, GPR; 98%, Merck Index-No. 011-002-00-6), and potassium hydroxide (KOH), as pellets (97%, BDH Laboratory Supplies, AnalaR; extra pure, Merck EC-Index-N.019-002-00-8), were utilized as activators.
Aluminum Chloride, AlCl3.6H2O (BDH Laboratory Supplies, GPR, extra pure, Merck EG-Nr./EC-No.201-705-8) was used to control the SiO2/Al2O3 ratio.
Ferric chloride anhydrous (iron (III) chloride), FeCl3 (ferrous chloride 96%; BDH Limited Poole England), chromium (III) chloride hexahydrate, CrCl3.6H2O (≥98.0%; Aldrich), and cadmium chloride, CdCl2.xH2O (98.0%; Aldrich) were applied for preparation of artificially polluted heavy metal aqueous solutions.
Starting material preparation for synthesis step
Sepiolite was crushed and sieved to the size of <200 mesh and then particles of <∼75 μm were selected for synthesis experiments. Subsequently, NaOH and KOH pellets were powdered and then added to the ground starting material (starting material/alkaline activator in 1:1.2 by weight proportion), and then put in a temperature-controlled furnace to prepare a fused precursor. Meanwhile, aluminum chloride solutions (1, 2, 3 N) were prepared by dissolving AlCl3.6H2O in distilled water on the basis of standard purification methods.
Methods
Starting material and synthetic products were characterized by using standard methods of phase and morphology determination.
Characterization methods
X-ray fluorescence spectroscopy
The bulk chemical composition of sepiolite was determined by x-ray fluorescence (XRF) on an Epsilon 3XLE PANalytical Energy–Dispersive Spectrometer equipped with a silver anode tube (max. voltage 50 kV, max. 3 mA, and max. tube power 15 W) and a high-resolution Si drift detector. The elemental analysis was performed on 10 mg of the raw material prepared through grinding by mortar and pestle and then firmly pressing and putting into the aluminum sample cup holder.
X-ray powder diffraction
The precise mineralogy and crystalline structure of both starting material and synthetic products were determined by means of x-ray diffraction (XRD), an Empyrean PANalytical X-ray diffractometer with Ni-filtered Cu-Kα radiation at 40 mA and 40 kV. The diffractometer was equipped with PIXcel1D detector. The Soller slits were 0.1 degree and an automatic divergence slit was used to irradiate 15 mm of the sample. Data collection was carried out in the 2θ range of 5–80°. The raw material and pure sodalites were ground to powder form (<15 μm) by a porcelain mortar and pestle and then mounted in aluminum plate sample holders.
Scanning electron microscopy
The morphology of raw material and synthetic products was characterized by scanning electron microscope (SEM), a ZEISS EVO50 Scanning Electron Microscope, in applying 10 kV and 20 kV extra high tension voltage, 7.0 mm and 8.5 mm initial working distance, and SE1 and VPSE detectors. The specimens were prepared by spraying dried raw material or product powder onto 12 mm aluminum stubs by using double-sided adhesive carbon discs. The specimens were then gold coated utilizing an Au-Pd Sputter Coater- Emscope SC500 to accurately image their surface.
Fourier transform infrared spectroscopy
The framework quality of starting material and synthetic products was identified by FTIR making use of a Bruker ALPHA spectrometer equipped with the high sensitivity DLATGS detector. The measurements were carried out through platinum-attenuated total reflection (ATR) single reflection module. The FTIR spectra were recorded within the region of 4,000–400 cm−1. Meanwhile, OPUS/Mentor software was used for evaluation of the results. The amount 10–15 mg of powdered samples (<15 μm) was placed onto the diamond ATR crystal surface. Then a force applied to the sample by the usage of one finger clamp mechanism for spectrum collection.
Inductively coupled plasma-optical emission spectrometry
The concentrations of heavy metal ions in the final solutions was determined by inductively coupled plasma-optical emission spectrometry (ICP-OES), utilizing an Agilent 5100 spectrometer equipped with Vista Chip II CCD detector, and solid-state radio frequency generator operating at 27 MHz. The measurements were simultaneously performed over 167 to 785 nm wavelength range. For ICP-OES metal analysis, the standard metal solutions were prepared to detect the amounts of Fe3+, Cr3+, and Cd2+ ion concentrations, which are present in the solutions before and after the exchange. The volume of exchanged solutions provided for the metal analysis was 10 mL in 15 mL centrifuge vial.
Synthesis method
Alkaline fusion method
Alkaline fusion method was applied before the hydrothermal treatment because of its critical role in enhancing hydrothermal conditions for dissolving aluminosilicates and synthesis of zeolites (Ríos et al., 2009). Moreover, in this procedure, some of the inert crystalline phases in the raw materials can fully react and yield products with high purity. In these experiments, which were performed in the chemistry laboratory of the school of applied sciences, University of Wolverhampton, powdered alkaline reagent (NaOH, or KOH) was added to the ground raw material to provide a fused mixture. Explaining in greater detail, at first, 6 g of crushed sepiolite was dry mixed with powdered NaOH or KOH (raw material/alkaline activator in 1:1.2 weight proportion). Furthermore, the resulting mixture was put in a ceramic crucible and fused at 650°C for 90 min in a temperature-controlled furnace. Then, the fused product was ground by a ceramic mortar and pestle and then the prepared powder was dissolved in distilled water for 30 min under stirring conditions. In the next step, aluminum chloride solution was added and the mixture was stirred for another 30 min to obtain a homogeneous precursor. Afterward, the precursor was poured in an autoclave and put in oven for 24, 48, 72 and 96 h, at temperatures of 100°C, 140°C, 180°C, and 220°C. Subsequently, after the certain period of time, the autoclave was removed from the oven, and was quenched in cold water to stop the reaction. The obtained mixture was filtered and rinsed several times with distilled water. After drying at 80°C overnight, the sample was powdered and prepared for further characterizations. A flowchart showing the processing of the primary material into synthesis products is presented in Fig. 1.

Processing of the raw material into synthetic products (Kamyab, 2020).
Batch-type adsorptive experiments
A series of batch adsorption experiments was carried out for evaluation of the adsorptive properties of pure synthesized sodalities. The influential parameters of contact time and initial ionic concentration were considered to be scrutinized in pursuance of their impact on the adsorption efficiency of the pure sodalites. Approximately 0.001, 0.01, and 0.001 M monometallic solutions of Fe, Cr, and Cd were prepared to investigate the adsorption efficiency of pure sodalities within various contact times of 1, 2, 4, 8, 12, and 24 h. The batch-type reactions were performed in glass containers with a volume capacity of 25 mL at room temperature, providing a given dosage of the pure sodalites as sorbents (sorbent/solution: 0.25 g/15 mL). The suspensions were kept in a rotary shaker with an agitation speed of 30 rpm for a given time interval. Each sorbent/solution sample was placed separately in every container, and at the scheduled time each container was removed from the shaker. Furthermore, the suspension was filtered and pH and electrical conductivity were measured. Eventually, the resultant filtrate was kept in a refrigerator at 4°C for further analysis.
The removal percentage of heavy metals was determined by Equation (1):
where Ce (mg/L) and C0 (mg/L) are equilibrium concentration and initial concentration of adsorbate in the solution, respectively.
Results
The bulk chemical composition of sepiolite, raw material, characterized by XRF analysis is shown in Table 2.
Chemical Composition of Sepiolite Sample Determined by X-Ray Fluorescence
LOI, loss on ignition.
Alkaline fusion products synthesized by the application of NaOH activator
Products formed by the alkaline fusion method by using NaOH activator are listed in Table 3.
Products Synthesized with Varying Amounts of NaOH and AlCl3·6H2O
Brc, brucite; Cal, calcite; Hl, halite; Mag, magnetite; MgCal, magnesium calcite; Sdl, sodalite; Zeo A, zeolite A.
Alkaline fusion products formed by the usage of KOH activator
Synthetic phases produced through the alkaline fusion method making use of KOH activator are listed in Table 4.
Products Synthesized Applying Varying Amounts of KOH and AlCl3·6H2O
Ame, amesite; Bay, bayerite; Cal, calcite; MgCal, magnesium calcite; Qz, quartz; Sdl, sodalite; Zeo A, zeolite A.
The results obtained from XRD analysis of the raw material and pure zeolite phases are shown in Fig. 2.

X-ray patterns:
The SEM images (Fig. 3) illustrate the morphology of sepiolite raw material, and both pure sodalite phases formed by the application of NaOH and KOH activators.
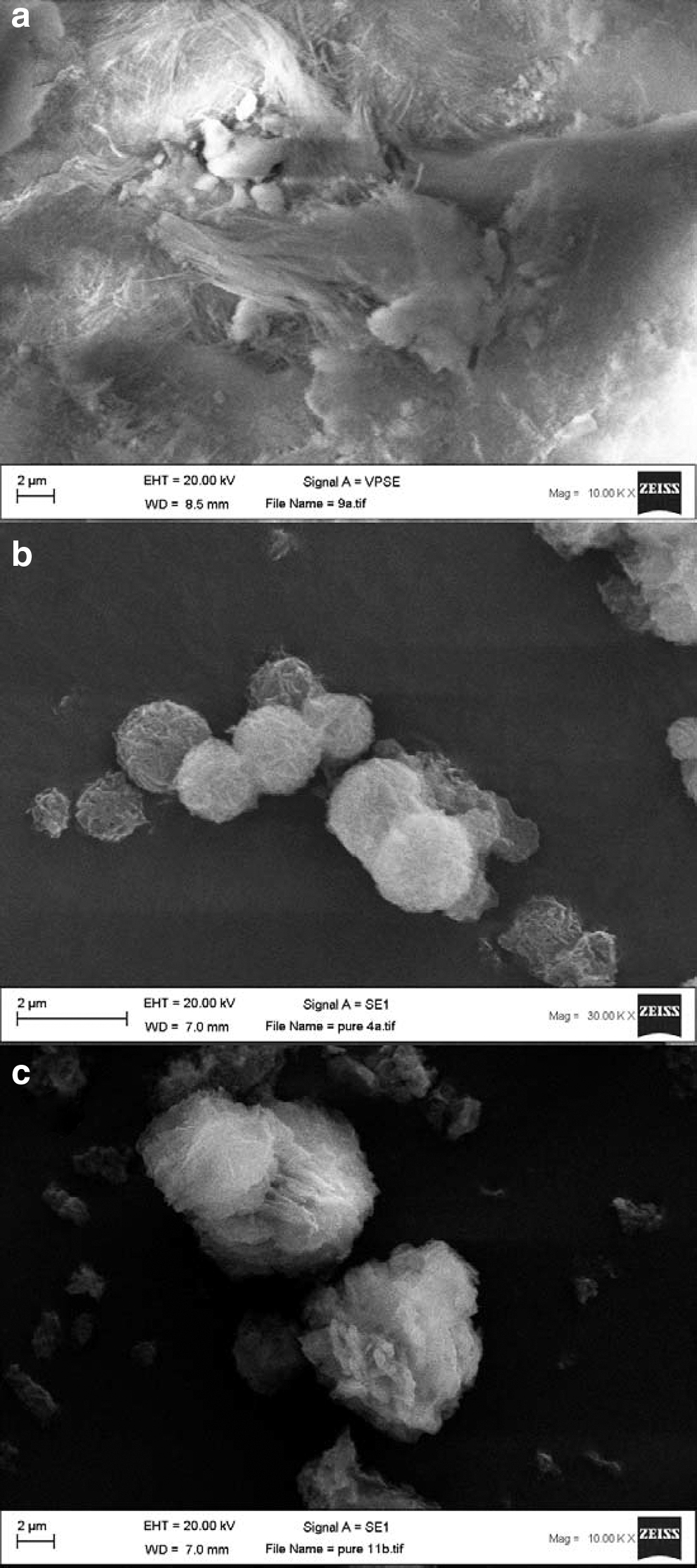
SEM images:
Infrared spectra for sepiolite and those of pure sodalite phases are presented in Fig. 4.
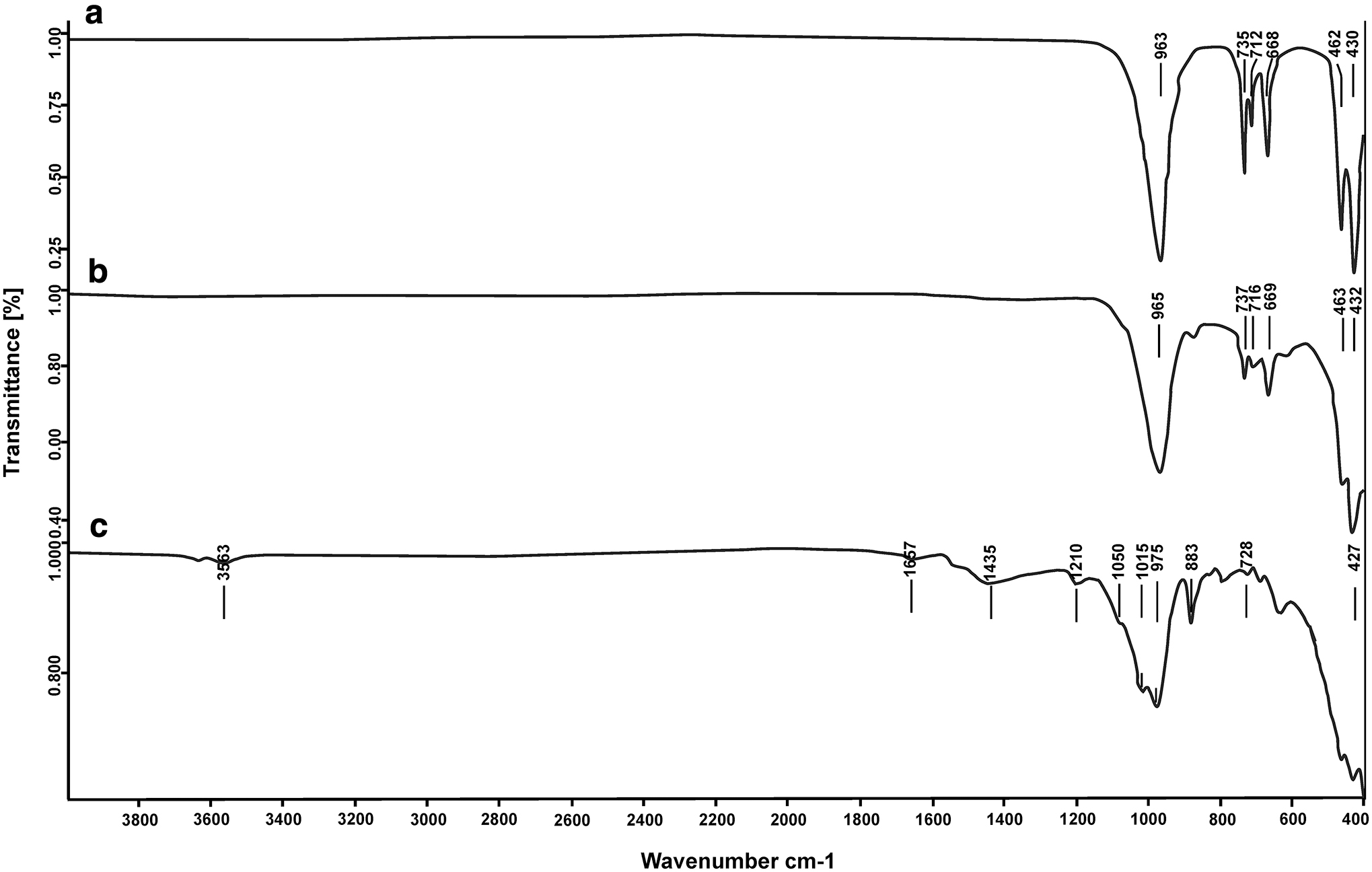
Infrared spectra:
Batch experiment results, including adsorption percentages and selectivity orders for Fe, Cr, and Cd removal, are reported in Tables 5 and 6.
Removal Percent of Fe, Cr, and Cd
Sdl 1, Sodalite 1; Sdl 2, Sodalite 2.
Fe, Cr, and Cd Removal Selectivity Orders
Sdl 1, Sodalite 1; Sdl 2, Sodalite 2.
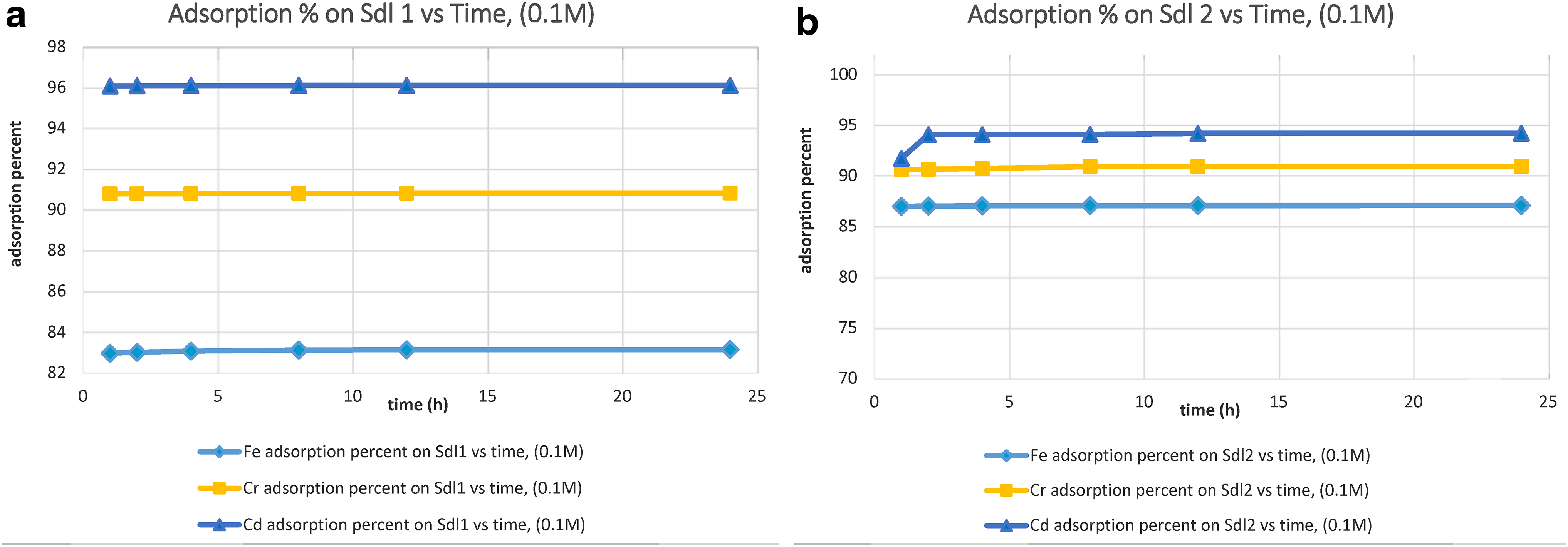
The removal percent of Fe3+, Cr3+, and Ni2+ ions from monometallic solutions is depicted in Figs. 5–7.
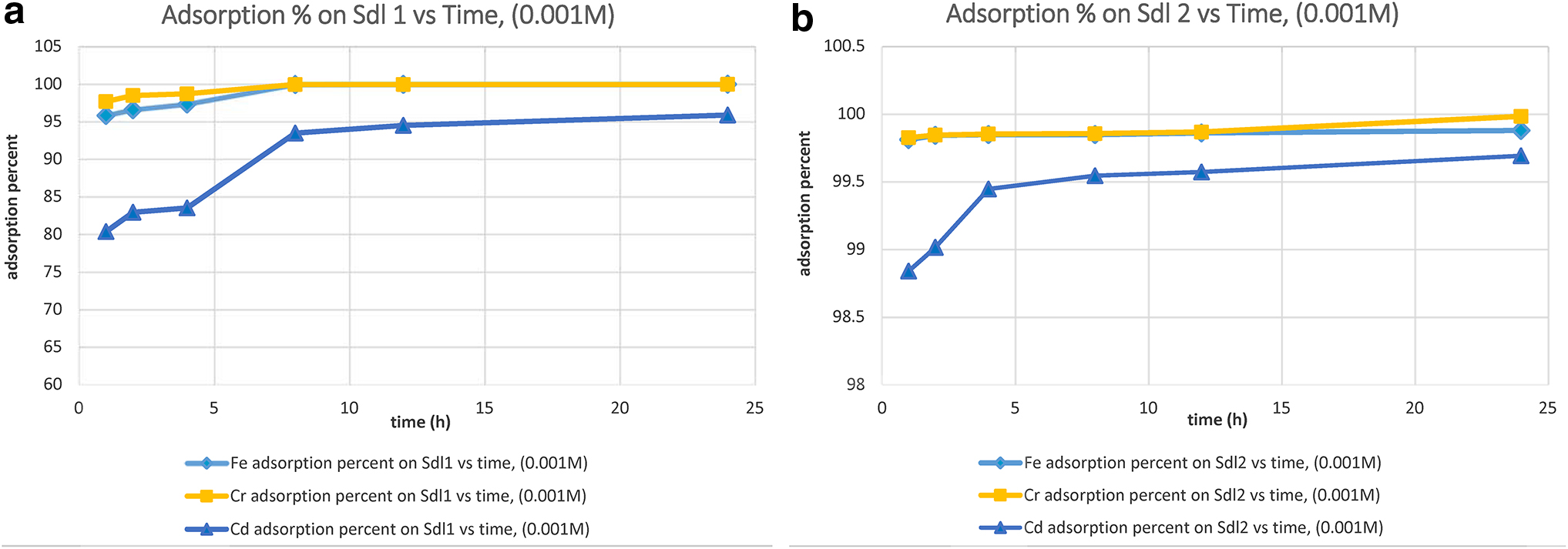
Adsorption percent on
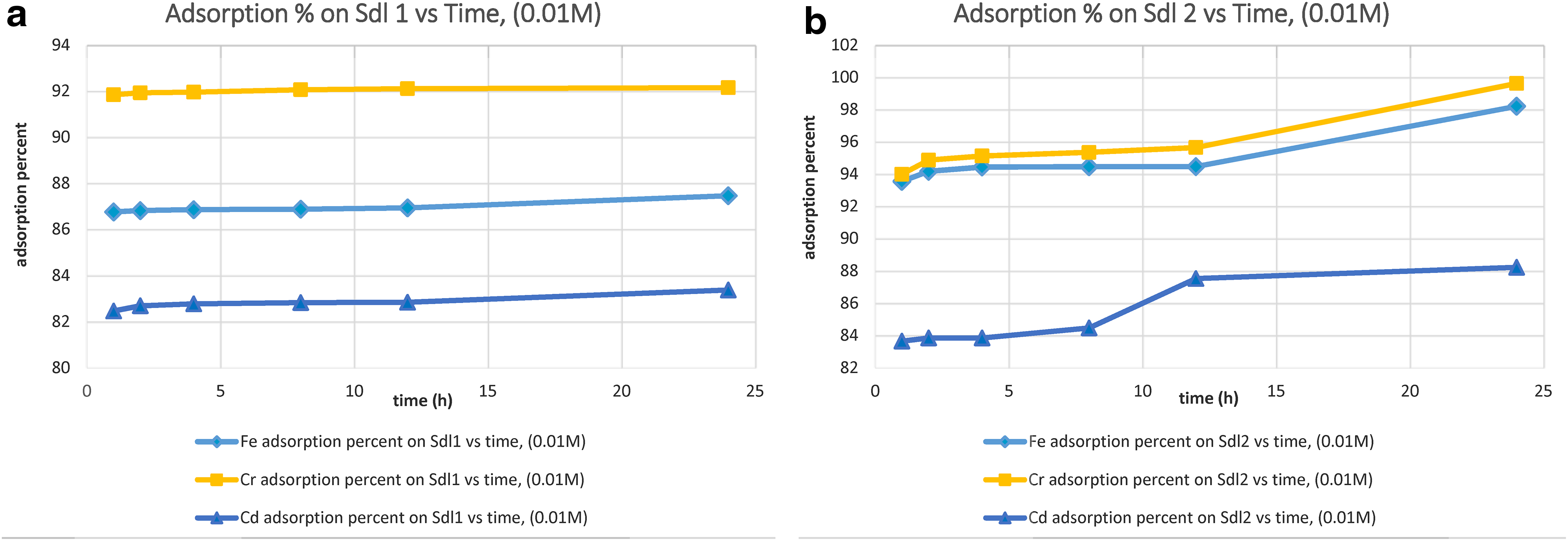
Adsorption percent on
Adsorption percent on
Discussion
Zeolites are usually synthesized in the presence of an amorphous gel phase. The solubility of this gel phase depends on the alkalinity. Thus, the alkalinity is an important parameter that controls the type of crystallizing zeolites as products of synthesis process (Robson, 1998). Moreover, it determines compositions of the synthetic zeolite phases.
Alkaline fusion products
The experimental studies in this research rely on the alkaline fusion approach by the application of powdered NaOH. According to Table 2, pure sodalite was synthesized successfully using 7.72 g powdered NaOH, at 140°C.
XRD patterns show pure products (Fig. 2) in samples synthesized at both 140°C and 180°C, with NaOH and KOH activators, for reaction time of 48 and 72 h, respectively. On the basis of these results, sodalite can be synthesized in the presence of low to medium amount of NaOH powder, 7.32–7.72 g, in the fused reaction gel, at 100–220°C. This reveals the importance of alkaline environment in zeolite formation (Sari et al., 2018). Meanwhile, from the theoretical point of view, higher alkalinity conditions imply high concentration of OH− in the experimental environment, which leads to higher solubility of Si and Al. High level of alkalinity destabilizes the Si-Al bonds in an aluminosilicate framework and correspondingly, a small yield of sodalite in the synthesized product is expected. Indeed, sodalite was not crystallized in high alkaline condition and in the presence of high OH concentration. On this matter, Lou et al. (2016) synthesized sodalite through alkali fusion method followed by a desilisication hydrothermal process by use of fly ash as starting material. According to their results, sodalite with high crystallinity was prepared at the basicity of 2 M, crystallization duration of 12 h, and crystallization temperature of 90°C.
In this study, KOH was also used as an activator in the fusion method to evaluate the possibility of zeolite formation under such conditions. As a result, pure sodalite formed making the use of 9.2 g KOH, at 180°C.
Therefore, the temperature in the alkaline fusion step may be also an effective parameter in the pursuit of pure phase formation. In the presence of KOH activator, pure sodalite was formed, at 180°C, for time duration of 72 h. Ideally, the composition of sodalite can be shown as Na8[AlSiO4]6(ClO4)2 (Borhade et al., 2012). Sodium is the main exchangeable cation in sodalite structure, balancing the charge of the aluminosilicate lattice (Franus et al., 2014). With this in view, K+ ion existence in synthesis conditions brings about formation of K-substituted sodalite (Rabiller et al., 2004). This can be elucidated through substitution of K+ in sodalite structure referring to M, which represents the monovalent cations like Na, Li, and K in the structural formula of sodalite, M8[AlSiO4]6.(XO4)2 (Borhade et al., 2012).
The SEM images in this work showed fiber bunches of sepiolite as primary material (Fig. 3a) with an intermediate length of fibers (in accordance with the fiber length classification (Del Río et al., 2011), In addition, the morphological aspects of pure synthesized sodalites produced by using activators of NaOH (Fig. 3b) and KOH (Fig. 3c) were illustrated through these microscopic images. Franus et al. (2014), in a study on synthesis of pure sodalite from fly ash under hydrothermal conditions showed the formation of spherical pure sodalite similar to desert roses. Moreover, Li et al. (2015) reported rhombic dodecahedron particles of sodalite with smooth surfaces synthesized from alkaline fused kaolinite. Furthermore, Shirani Lapari et al. (2015) reported the formation of a sodalite with regular spherical morphology, which was formed under the influence of organosilane through hydrothermal reactions.
The results concerning FTIR spectrum of pure sodalite synthesized in applying NaOH activator (Fig. 4a) revealed absorption bands at 430 and 462 cm−1 stemming from O−T−O bending vibration. In addition, T−O−T (T = Si, Al) symmetric stretching vibrations were displayed at 668, 712, and 735 cm−1 bands, while, the absorption band at 963 cm−1 was ascribed to asymmetric stretching vibration. According to Inada et al. (2005), the band assigned to the T−O−T asymmetric stretching was seen at 997 cm−1, absorption bands at 715 and 657 cm−1 were associated with the T−O−T symmetric stretch, and the bands observed at 459 and 430 cm−1 were related to bending vibration of O−T−O and bending vibration of single four-membered ring of sodalite, respectively.
The infrared spectrum of pure sodalite synthesized in the presence of KOH activator (Fig. 4b) exonerated the presence of bands at 432 and 463 cm−1 resulting from O−T−O bending vibrations. Besides, the T−O−T symmetric stretching vibrations were displayed at 669, 716, and 737 cm−1 absorption bands. Further to this, absorption band at 965 cm−1 was related to asymmetric stretching vibrations. On this matter, the FTIR spectrum of a gallosilicate perchlorate sodalite, for instance, demonstrated three sets of framework modes: an absorption band for T−O−T (T = Ga or Si) asymmetric stretching vibration at 955 cm−1, two absorption bands for T−O−T symmetric stretching at 534, and 621 cm−1, and two absorption bands for O−T−O bending vibration at 343 and 451 cm−1 (Borhade et al., 2010). Indeed, positions of symmetric and asymmetric T−O−T modes are dependent on the framework expansion (Henderson and Taylor, 1979). In other terms, any shift in the IR frequency as a function of cation exchange comes from the framework expansion, the expansion that can be verified on the basis of cell constants (Jin et al., 2008; Borhade et al., 2012).
The FTIR spectrum of sepiolite raw material (Fig. 4c) that displayed an absorption band at 3,563 cm−1 relating to OH groups in the octahedral sheet of sepiolite framework, which as reported by Frost et al. (2001) and Chahi et al. (2002), originates from OH stretching vibration in the surface of sepiolite. The absorption band observed at 1,657 cm−1 was ascribed to H−O−H bending vibrations representing the zeolitic water in the channels and bound water coordinating to magnesium in the octahedral sheet. In light of this, Farmer (1974) verified the existence of two partially resolved peaks at 1,630 and 1,656 cm−1 associated with the adsorbed and zeolitic water, respectively. Absorption band at 1,435 cm−1 was related to calcite impurity in sepiolite raw material. The Si−O coordination bands at 1,210, 1,050, and 1,015 cm−1 represent the stretching of Si−O in the Si−O−Si groups of the tetrahedral sheet. The absorption region appearing at 728, 883, and 975 cm−1 was ascribed to T−O−T (T = Al, Si) symmetric stretching vibrations and correspond to the band characteristics of silicate minerals, while, the absorption band at 427 cm−1 was associated with O−T−O bending vibration. Based on the FTIR analysis performed by Lazarević et al. (2007) on a sample of sepiolite, the absorption bands disclosed at 3,690, 3,568, and 3,422 cm−1 were related to the vibration of inside Mg-OH stretching, coordinated water (edge Mg-OH stretching), and zeolitic water, respectively. The band observed at 1,671 cm−1 was due to the vibration of zeolitic water, and bands centered at 1,016 and 460 cm−1 were ascribed to Si−O−Si vibration. Furthermore, the bands seen at 1,215, 1,076 and 980 cm−1 were associated with Si−O bonds and the band revealed at 437 cm−1 was assigned to octahedral-tetrahedral bonds (Si−O−Mg bonds), while, the bands displayed at 690 and 637 cm−1 correspond to Mg-OH bond vibrations.
Adsorption experiment result evaluation
The ICP-OES results for final leachates (Tables 5 and 6 and Figs. 5–7) indicated that for 0.001 M solutions, pure sodalite 1, which was synthesized by NaOH activator at 140°C, removed Fe3+ and Cr3+ ions at the highest adsorption amounts with an increasing rate between 1 and 2 h, and then with a constant rate at equal amounts up to 24 h. This pure sodalite adsorbed Cd2+ ion at the highest amounts of adsorption with a rate between 1 and 2 h, and then with an increasing continuous rate between 4–8 and 24 h, whereas pure sodalite 2 synthesized by KOH activator, at 180°C, had the highest level of adsorption for Fe3+ and Cr3+ ions between less than 1 and 24 h. The latter pure sodalite removed Cd2+ ion increasingly at the highest adsorption amounts between 1 and 4 h, and then adsorption steadily increased up to 24 h. The cation uptake percent within 1–24 h was as follows: pure sodalite 1 adsorbed Fe3+ between 95.7% and 99.9%, Cr3+ from 97.6% to 99.9%, and Cd2+ within the range of 80.3–95.8%, while pure sodalite 2 cleaned up Fe3+ with percentages changing between 99.85 and 99.87%, Cr3+ from 99.8% to 99.9%, and Cd2+ within the range of 98.8–99.6%. In sense of cation selectivity orders, both pure sodalites 1 and 2 adsorbed Fe3+ and Cr3+ at the highest amounts and Cd2+with less amounts than the former cations, during 1–24 h.
For 0.01 M solutions, pure sodalite 1 removed Fe3+ and Cr3+ ions constantly at the highest amounts between 1 and 24 h. This sodalite cleaned up Cd2+ ion incessantly at the maximum amount to 2 h and then with an inalterable rate, keeping the same amounts, up to 24 h. However, pure sodalite 2 adsorbed Fe3+ and Cr3+ ions with an additive rate at the highest amounts of adsorption between 1 and 2 h, then continuously at the similar amounts up to 12, and then to 24 h. This synthesized sodalite adsorbed Cd2+ ion with a persistent rate between 1 and 8 h, and with a growing rate at the maximum adsorption amount to 12 h, subsequently with a permanent rate up to 24 h. In compliance with the removal percentages of cations measured during 1 to 24 h, pure sodalite 1 adsorbed Fe3+ within the range of 86.7–87.4%, Cr3+ between 91.8% and 92.1%, and Cd2+ from 82.4% to 83.3%, while, pure sodalite 2 cleaned up Fe3+ between 93.5% and 98.2%, Cr3+ from 93.9% to 99.6%, and Cd2+ within the range of 83.6–88.2%. In relationship to cation selectivity orders, both pure sodalites 1 and 2 removed Cr3+ and Fe3+ at the highest amounts and Cd2+ with the amounts less than those of Fe3+ and Cr3+, within 1–24 h.
Under the influence of 0.1 M solutions, pure sodalite 1 adsorbed Fe3+ ion at the highest amounts between 1 to 24 h and cleaned up Cr3+ and Cd2+ ions continuously at the maximum amount between 1 and 24 h. Conversely, pure sodalite 2 removed Fe3+ and Cr3+ ions progressively at the highest amounts between 1 and 24 h and adsorbed Cd2+ ion with a growing rate at the highest level to 2 h, and then persistently up to 24 h. The outcomes demonstrated the cation uptake percent, within 1–24 h, as follows: pure sodalite 1 adsorbed Fe3+ with the range of 82.9–83.1%, Cr3+ with the percentages changing from 90.7% to 90.8%, and Cd2+ between 96% and 96.1%. Contrarily, pure sodalite 2 removed Fe3+ with the percent ranging from 86.9% to 87%, Cr3+ with the percentages changing from 90.5% to 90.9%, and Cd2+ between 91.7% and 94.1%. In connection with the selectivity orders of adsorptive cations, during 1–24 h, pure sodalite 1 removed Cd2+ and Cr3+ at the highest amounts and Fe3+ with less amounts, while pure sodalite 2 adsorbed Cd2+at the highest amounts, and Cr3+ and Fe3+ with less amounts.
In sense of initial concentration, for both pure sodalite 1 and pure sodalite 2, the optimum adsorption amount was disclosed for Fe3+ and Cr3+ ions in 0.001 M solutions. Nevertheless, the maximum removal for Cd2+ ion was seen in 0.1 M solutions. With respect to contact time, the maximum amount of adsorption for 0.001 M solutions was observed between 1 and 2 h for pure sodalite 1. For pure sodalite 2, it was mostly between 30 min and 1 h. Under the effect of 0.01 and 0.1 M solutions, the highest level of adsorption was revealed between 30 min and 1 h, for both sodalites 1 and 2.
The parameters such as pH, temperature, concentration, and induction speed have drawn scientists' attentions in sense of their impacts on zeolite capability for heavy metal removal from aqueous solutions (Trgo and Perić, 2003; Perić et al., 2004; Salunkhe and Raut, 2012; Shaheen et al., 2012; Visa and Popa, 2015; Hesnawi et al., 2017).
The period of contact time was evaluated in this work, considering its effect on the adsorption progress. The results substantiated that the maximum adsorption of heavy metal ions can be seen early on, since the active sites on surface of the adsorbent were readily accessible, in the first 30 min. Nevertheless, as time elapsed, there was a decline in the adsorption rates coming from slower adsorption progress. This was assigned to more sites occupied through proceeding of the adsorption. That is to say, the impact of this parameter was elucidated by a rapid adsorption rate in the first 30 min followed by a gradual adsorption over time to obtain equilibrium. In actual fact, the equilibrium achieves through time due to the diminution in available active sites on the adsorbent (Mehdizadeh et al., 2014).
The initial concentration of metal ions seems to influence their uptake efficiency. So that, the initial ion concentration plays a significant role in the adsorption proceeding and can be regarded as a driving force to conquer the mass transfer residence between the liquid phase and solid phase. On that account, an accession in metal ion concentration causes increase of the metal uptake (Ziyath et al., 2011). Indeed, at low concentration of metal ions, the available adsorption sites can be easily occupied because the gradient between cations in solution and the number of available vacant sites on the adsorbent is low. Hence, adsorption progress is independent of the initial concentration when the latter is low enough. Conversely, at higher concentrations, the number of ions competing for available sites to bind on the surface of the adsorbent rises and subsequently reduces the number of binding sites. Meanwhile, the average distance between the adsorbed ions will be reduced, affecting the surface charge distribution (Quintelas et al., 2009). With this in view, the efficiency of adsorption is dependent on the initial concentration since the number of available sites is less than the number of existed metal ions. Therefore, an enhancement in the initial concentration brings about a drop in the removal rate (Horsfall et al., 2006). In this regard, as shown by Sallam et al. (2017), Cr (VI) removal capacity of polymer-clay composites decreased with increase in the initial Cr concentration, which was explained through quick occupation of the available binding sites by Cr, thus preventing further adsorption on the adsorbents.
The uptake efficiency of Fe3+, Cr2+, and Cd2+ ions might be demonstrated through the selectivity orders resulting from adsorption experiments performed by using sodalites 1 and 2, for periods of 1–24 h. So that, for 0.001 and 0.01 M solutions, the metal ions showed an order of Cr > Fe > Cd, while, a sequence of Cd > Cr > Fe was seen as the metal ions were taken out from 0.1 M solutions. In this context, the cationic exchange of zeolites can be inspected to vindicate the adsorption experiment outcomes. Principally, the zeolite's ability to exchange cations depends on the number of Al3+ ions that displace Si4+ in the zeolite tetrahedral framework (Mumpton, 1999; Ouki and Kavannagh, 1999). In this point, a higher content of Al induces more ions to replace exchangeable cations within the framework. In addition, zeolite attitude toward adsorption progression and their performance in cation exchange process are certainly influenced by the hydration size of metal ions (Zamzow et al., 1990). With that in mind, in the chemical adsorption process, the ions having larger ionic radii, and therefore smaller hydrated radii, are preferably adsorbed by the adsorbents. In contrast, ions with smaller ionic radii tend to remain in the aqueous solutions, since they possess larger hydrated radii (Zhang et al., 2011). This exonerates the highest adsorption level of Cd2+cations in 0.1 M aqueous solutions.
The overall mass transport of Fe3+, Cr3+, and Cd2+ cations in aqueous solutions can be illustrated in the following steps: (1) cation diffusion through the solutions up to sodalite particles, (2) cation diffusion through the active sites through exchange, (3) chemical exchange between the cations and exchangeable cations of Na+ and K+ at exchange site in sodalite (β cage), and (4) displaced cation diffusion out of sodalite framework (Ćurković et al., 2014). Accordingly, the metal ion uptake could be mainly attributed to the ion exchange process.
Conclusions
In a nutshell, as a raw material, sepiolite possesses potential for the production of pure zeolite phases (regardless of the type of applied activators), which subsequently can be used in industrial and environmental applications.
Synthesis of pure sodalites verified the expedience of alkaline fusion step being applied before hydrothermal reactions. This was achieved through evaluation of the alkaline medium and temperature impact on zeolite synthesis. On top of that, the optimal conditions for pure sodalite synthesis with the usage of both NaOH and KOH activators were determined.
Pure sodalite efficiency for heavy metal removal was scrutinized through batch adsorption experiments. Contact time and initial ionic concentration were important factors affecting the general reaction rate of adsorption. The outcomes confirmed pure sodalite suitability for Fe3+, Cr3+, and Cd2+ removal from aqueous solutions.
In applying 0.01 and 0.1 M solutions, it was found out that the optimum adsorption occurred between 30 min and 1 h, for both sodalites 1 and 2, whereas the maximum adsorption for pure sodalite 1 was between 1 and 2 h, for 0.001 M solutions. Pure sodalite 2 showed maximum adsorption between 30 min and 1 h for the same concentration.
Besides their potential as catalysts, these synthetic pure sodalities seem to be promising minerals for environmental applications, not least, because of their capability to get recovered after cation exchange processes.
Footnotes
Acknowledgments
First Author would like to express her gratitude to Professor Craig D. Williams, her research advisor, for providing the opportunity to use the research facilities in the chemistry laboratory at University of Wolverhampton. We really appreciate the administrative staff of the School of Applied Sciences of the University of Wolverhampton for all their help. We also acknowledge David Townrow, Diane Spencer, and Dr. Keith R. Jones for their assistance with the acquisition of XRF, XRD, FTIR, and SEM data, respectively.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.