
Abstract
Livestock manure and digestate (biogas slurry [BS] and biogas residue [BR]) contain bacteria that can potentially destroy the environment microecology and affect human health. This is a problem in China since it is a big producer of livestock and poultry. Thus, herein we have analyzed the bacterial content of both manure and digestate in 18 pig farms located in the Guangxi Zhuang Autonomous region. Specifically, we show that mid-temperature anaerobic fermentation affects both bacterial colony structure of swine manure (SM) and the relative abundance of bacteria and human pathogens. Indeed, the bacterial composition of SM changed after manure was fermented into BR and BS. These changes were dependent on bacterial adaptability; although some bacteria were reduced after fermentation, we observed an increase in Novosphingobium, Acidovorax, Longilinea, and Mycobacterium. The first three can potentially degrade polycyclic aromatic hydrocarbons and other harmful substances and may have a beneficial effect on the control of soil environmental pollution. However, the increased abundance of Mycobacterium poses a greater threat to human health. Thus, the transition from SM to digestate in the current condition for mid-temperature anaerobic fermentation needs to be optimized to reduce the entry of potential human pathogens in the soil after anaerobic fermentation.
Introduction
The livestock and poultry breeding industry, especially pig breeding, is the most important economic pillar of the Guangxi Zhuang Autonomous Region in China. However, in recent years, the pig industry in the Guangxi Zhuang Autonomous Region has restricted the development of its pig industry because of long-term environmental pollution caused by microbial contamination after treatment of swine manure (SM).
Indeed, the rapid development of animal husbandry leads to serious environmental problems, such as air, water, and fecal pollution, related to the rapid development of animal husbandry (Wang et al., 2017; Bird et al., 2019; Otte et al., 2019; Yu et al., 2019). Manure pollution causes bacterial contamination that can affect human health (Garcia et al., 2014; Hatvani et al., 2018; Katarzyte et al., 2018; Nshimyimana et al., 2018; Kapembo et al., 2019; Karkman et al., 2019; Toribio-Avedillo et al., 2019). Thus, to improve the urban environment and to ensure public health safety, it is urgent to improve excrement treatment.
In modern animal husbandry, livestock manure is often returned to the soil as fertilizer after fermentation, a process that produces energy in the form of methane (Liu et al., 2013; Paranhos et al., 2020). Thus, the soil becomes infected with zoonotic bacteria, and these may use plants as a vector to later infect humans that consume plant products as food. Also, zoonotic bacteria present in the soil can infect rural people that live close to farmland through aerosol (Muscatello et al., 2009; Gao et al., 2015).
It is known that fermentation changes the composition of the livestock manure and the original microbial community in the soil environment. For example, it increases the proportion of Firmicutes, Proteobacteria, and Zygomycota that are known to prefer a nutrient-rich environment (Davide et al., 2016). These changes affect the general ecological balance and can change the ability of the soil to degrade organic pollution, for example, chlorimuron ethyl (Li et al., 2017) or conversion of heavy metals like lead (Liu et al., 2017).
Various pathogens have been identified in farms after the return of livestock manure to the field, for example, Salmonella sp (Lynch et al., 2018), Escherichia coli O157:H7 (Tanaro et al., 2018), Campylobacter jejuni, Yersinia enterocolitica (Bockemuhl and Roth, 1978), or Clostridium perfringens (Schafer et al., 2013) were the main pathogens in farms. Salmonella is an the important pathogen present in the environment and including more than 200 serotypes (Venglovsky et al., 2006).
The microorganism composition in livestock and poultry manure and consequently the structure of the resulting microbial flora after mid-temperature anaerobic fermentation depends on the feeding methods used and on the living environment. Therefore, studies must be performed across different regions. Fermentation producer should be improved by producing characteristic lineages of microbial changes.
These studies are specially relevant for the Guangxi region in China due to the importance of the pig industry there. Thus, in the present study, we have performed a high-throughput sequencing analysis of SM and digestate after mid-temperature anaerobic fermentation in samples collected from 18 pig farms in Guangxi Zhuang Autonomous region. The result obtained is an important reference for optimization and harmless treatment of livestock manure and for the rational use of digestate to control microbial contamination.
Materials and Methods
Sample collection and DNA extraction
Samples were obtained from 18 pig farms located in the Jiuzhou river basin, in the Guangxi Zhuang Autonomous region in Southern China: 15 samples of SM, 16 samples of biogas slurry (BS), and 17 samples of biogas residue (BR). In all these farms, manure was treated using the mid-temperature anaerobic fermentation method for 7 days, and both BS and BR samples were freshly obtained after this period. Samples were taken from upper, middle, and lower layers and used as independent samples (Supplementary Table S1). Samples were frozen immediately under liquid nitrogen, send to the laboratory, and stored at −20°C until further use. Physical and chemical properties of stool samples (Supplementary Table S2) were tested based on the Chinese national standard (GB/T 25169-2010).
DNA from samples was extracted using the FAST DNA Kit following the manufacturer's instructions. The integrity of the extracted DNA was tested using agarose gel electrophoresis, and subsequent experiments were performed immediately.
16S rRNA gene amplification and MiSeq sequencing
The DNA for polymerase chain reaction (PCR) amplification was quantified using the Qubit3.0 DNA Tested Kit before PCR amplification to determine the amount of DNA template to include. The universal V3-V4 variable region primers on 16S rDNA were the 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAAT CC-3′). Barcode and adapter were incorporated between the adapter and the forward primers.
PCR amplification was performed in two rounds. The first used 2 × Taq Master Mix (Vazyme), 30 μL reaction systems. The PCR reaction was performed in T100™ Thermal Cycler (Bio-Rad) in a solution containing 15 μL of 2 × Taq Master Mix, 1 μL of 10 μM Bar-PCR primer F, 1 μL of 10 μM primer R, 10–20 ng of genomic DNA, and sterile double-distilled H2O in a total volume of 30 μL. The process consisted of an initial 3 min denaturation step at 94°C for 3 min, followed by 5 cycles of denaturation at 94°C for 30 s, and annealing at 45°C for 20 s, with an extension at 65°C for 30 s. This was followed by 20 cycles comprising denaturation at 94°C for 20 s, and annealing at 55°C for 20 s, with an extension at 72°C for 30 s were continuing. Finally, a total extension of 72°C for 10 min was carried out. The PCR product was kept at 10°C for a second round of amplification used to add Illumina bridge PCR-compatible primers. In this second round of PCR amplification, we used 15 μL of 2 × Taq Master Mix, 1 μL of 10 μM primer F, 1 μL of 10 μM primer R, 20 ng product from the last round of PCR, and sterile double-distilled H2O in a total volume of 30 μL. This second round of amplification was simpler than the first and included initial 3 min denaturation at 95°C, five cycles of denaturation at 94°C for 20 s, annealing at 55°C for 20 s, an extension at 72°C for 30 s, and finally an extension at 72°C for 5 min.
PCR products were observed with 2% agarose gel electrophoresis. After the detection, PCR products were purified and recovered using Agencourt AMPure XP. Quantification of PCR products was performed using the Qubit3.0 DNA Detection Kit. DNA and sequencing reagent were mixed in a 1:1 ratio and sequenced by MiSeq.
Sequence data analysis
The original image data files from Illumina Miseq sequencing were base calling by CASAVA and translated to sequenced reads, that is, raw reads. The quality of the original data was assessed by Prinseq (Version 0.20.4) to get effective data and the following study was based on effective data. Sequences were clustered into different operational taxonomic units (OTUs) by Usearch (Version 5.2.236). A similarity level of 97% for OTU was usually used for bioinformatics analysis.
The microbial richness, diversity, and coverage were described by the Chao1, Shannon–Wiener diversity index, and Good's coverage indices, respectively, using Alpha diversity calculated by Mothur (Version 1.30.1). The Rarefaction Curve was drawn by Mothur and R according to OTU at 97% similarity. Taxonomic annotation of the individual OTUs was based on representative sequences using RDP's 16S Classifer, Version 2.12 (Wang et al., 2007) at a threshold value of 80%. Based on the results of species classification, the community structure of these samples with different microbial classification levels was analyzed by statistical methods.
Statistical analysis
Based on the relative abundance of bacteria and potential human pathogens in samples, the analysis of similarities (ANOSIM) of nonparametric multivariate analysis was used to determine the significance of sampling grouping. Significance in grouping was determined by R > 0 and p < 0.05. The relative abundances of major bacteria and major human pathogens in SM, BS, and BR were analyzed using the Kruskal–Wallis and Bonferroni tests.
Accession numbers for the sequencing data
The sequence information was deposited in the NCBI Short Read Archive database under accession nos. SRR12151389–SRR12151436.
Results
High-throughput sequencing results and microbial diversity
A total of 48 samples in SM, BS, and BR were analyzed by high-throughput sequencing (see Supplementary Table S1 for information on grouping and sampling spots). Illumina Miseq sequencing generated 1,735,167 high-quality microbial sequences with an average of 36,149 sequences per sample. The Good's coverage ranged from 88% to 99%. Chao1, ACE, and Shannon and Simpson indices reflect the species richness and diversity (Fig. 1). The latter were higher in BR and lower in SM and BS, with significant differences in the same types of sampling spots. With a similar level of 97%, sequences were grouped into the same OTU. We obtained 143,148 OTUs, with large variations in the number of OTUs in each type of sampling points, possibly due to differences in species richness. The number of OTUs obtained at each point in each sample class (Supplementary Table S3) was also very unusual, possibly due to geographical reasons.
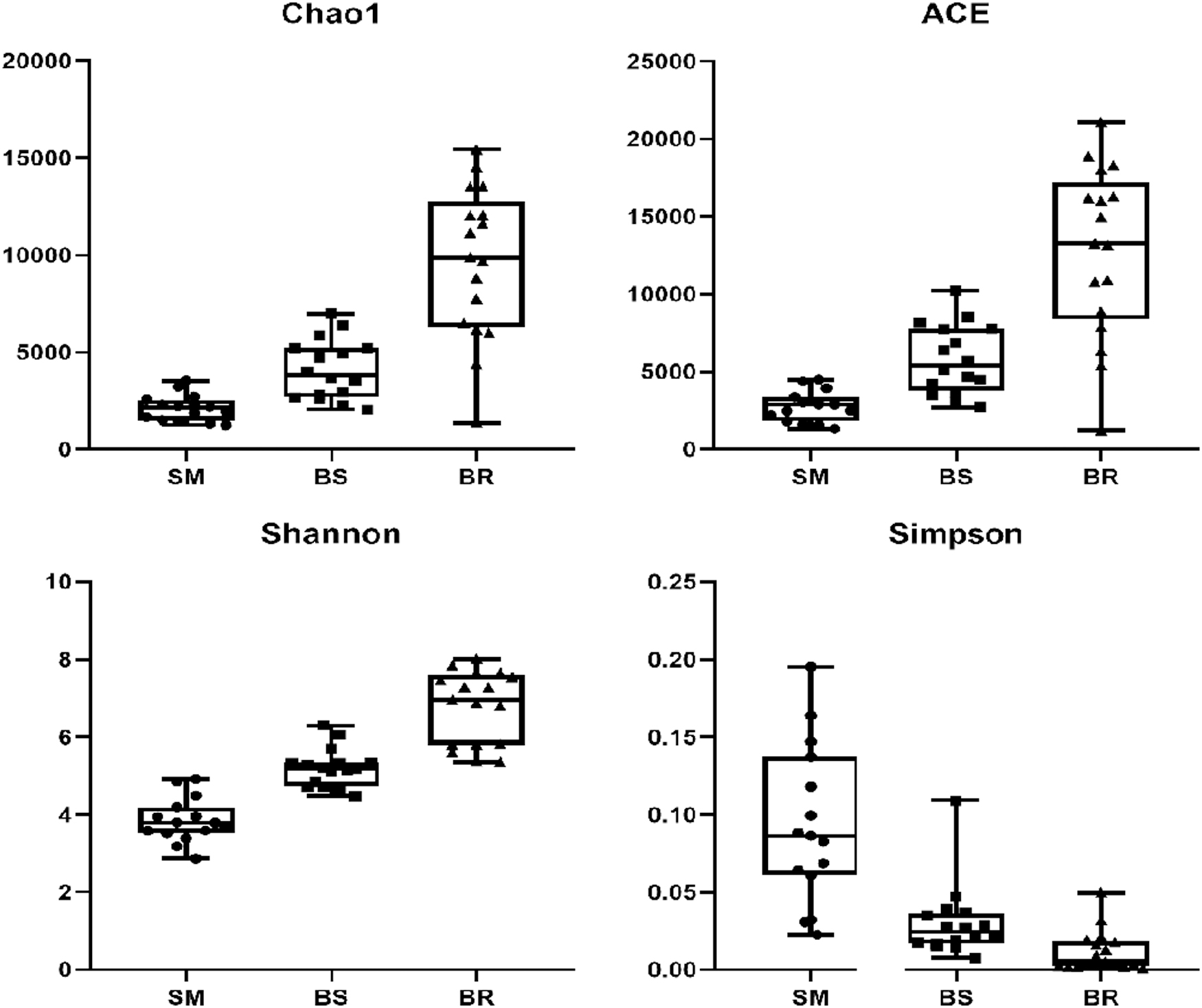
Microbial diversity index in SM, BS, and BR samples. BR, biogas residue; BS, biogas slurry; SM, swine manure.
Composition of bacteria in SM and digestate
In SM, Actinomycete (0.13–29.89%), Bacteroides (1.09–33.67%), Firmicutes (4.91–88.71%), and Proteobacteria (0.49–48.52%) accounted for more than 88% of all microorganisms. Firmicutes was the main phylum and its presence varied greatly among different samples. In BS, Actinomycetes (2.02–39.42%), Bacteroides (8.38–21.44%), Firmicutes (0.56–49.56%), and Proteobacteria (32.24–58.3%) accounted for more than 78% of all microorganisms. In this case, the main bacterial group was Proteobacteria, reflecting the changes in the structure of the microorganisms in SM after mid-temperature anaerobic fermentation. Finally, in SS, Bacteroides (1.76–16%), Chloroflexi (1.62–22.81%), Firmicutes (6.67–49.76%), and Proteobacteria (7.19–43.89%) constituted 58% of all microorganisms. Different media in BS and BR may lead to differences both in bacterial species and in abundance.
At the genus level, detection threshold was set at 1% of the relative microbial abundance, and detection rate >50% was considered high. In SM samples, Clostridium sensu stricto (86.67%), Terrisporobacter (86.67%), Lactobacillus (80%), and Streptococcus (73.33%) were those with higher detection rates (Fig. 2). In BS samples, those were Flavobacterium (93.75%), Novosphingobium (68.75%), Acidovorax (68.75%), Terrisporobacter (62.5%), Clostridium sensu stricto (56.25%), Mycobacterium (56.25%), and Acinetobacter (50%). Finally, in BR samples, the most easily detected were Clostridium sensu stricto (100%), Terrisporobacter (76.47%), Ornatilinea (64.71%), and Longilinea (58.82%).
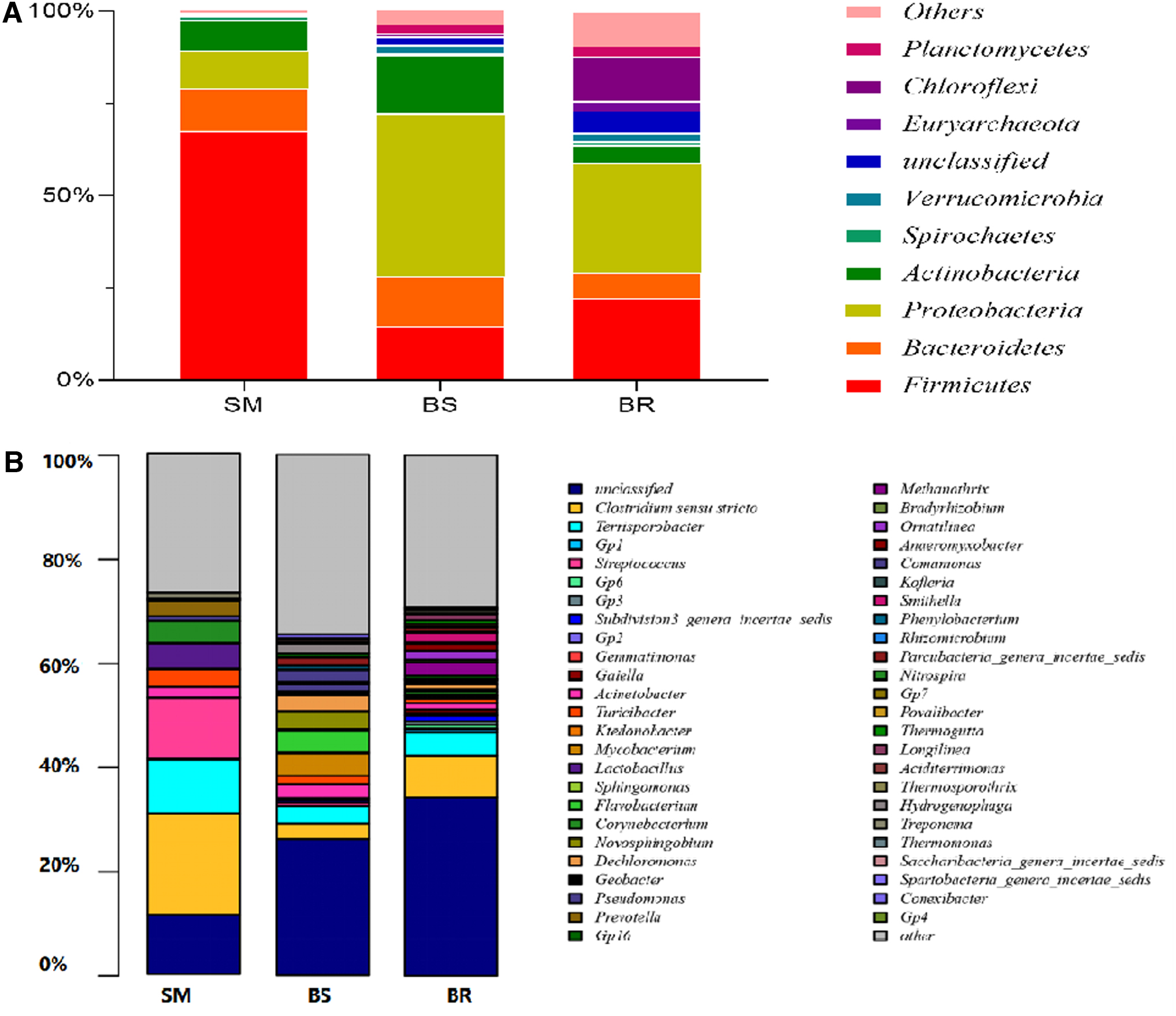
Composition and abundance of bacteria at the phylum level
Another important threshold to screen bacteria with higher relative abundance was the median relative abundance, which was set at 1%. In SM samples, this condition was met by Clostridium sensu stricto (20.73%), Streptococcus (10.09%), Terrisporobacter (5.45%), and Lactobacillus (2.67%). In BS samples, this condition was met by Flavobacterium (3.595%), Novosphingobium (2.37%), Clostridium sensu stricto (1.41%), Terrisporobacter (1.33%), Mycobacterium (1.31%), Acinetobacter (1.26%), and Acidovorax (1.22%). Finally in BR samples, those with higher relative abundance were Clostridium sensu stricto (5.54%), Terrisporobacter (2.96%), Ornatilinea (1.83%), and Logilinea (1.07%).
The detection rate and the median of relative abundance of bacteria in SM, BS, and BR were statistically correlated according to the Spearman test (p < 0.01). That is, bacteria with higher detection rate was generally of higher relative abundance and bacteria with these properties was considered as major bacteria. According to this, and using the detection rate of 50% as the threshold combined with the relative abundance data, Clostridium sensu stricto, Lactobacillus Streptococcus, and Terrisporobacter were the major bacteria in SM samples. Acinetobacter, Acidovorax, Clostridium sensu stricto, Flavobacterium, Mycobacterium, Novosphingobium, and Terrisporobacter were the major bacteria in BS samples. Finally, Clostridium sensu stricto, Longilinea, Ornatilinea, and Terrisporobacter were the main bacteria in BR samples.
Occurrence and distribution of potential human pathogens
The VFDB database contains a total of 32 potential human pathogens (Supplementary Table S3). The 16S rDNA sequencing technology used in this work could annotate 26 potential human pathogens. In this part of the analysis, detection was defined as absolute abundance of potential human pathogens >0%. In SM, BS, and BR samples, we detected 14, 21, and 17 human pathogens, respectively.
Potential human pathogens with detection rate ≥80% (the percentage of the samples with relative abundance >0% versus the total number of samples in the same group) were regarded as having high detection rate. In SM samples, those were Corynebacterium (93.33%), Enterococcus (93.33%), Streptococcus (93.33%), Acinetobacter (86.67%), and Pseudomonas (80%). In BS samples, these were Acinetobacter (100%), Aeromonas (100%), Mycobacterium (100%), Pseudomonas (100%), Streptococcus (87.5%), Bordetella (87.5%), Bacillus (81.25%), Corynebacterium (81.25%), and Enterococcus (81.25%). In BR samples, these were Acinetobacter (100%), Bacillus (100%), Mycobacterium (100%), Pseudomonas (100%), and Corynebacterium (88.24%). The potential human pathogens in SM, BS, and BR samples (Fig. 3) were mainly Firmicutes (Enterococcus, Streptococcus, Bacillus), Proteobacteria (Acinetobacter, Pseudomonas, Aermonas, and Bordetella), and Actinomycetes (Corynebacterium and Mycobacterium).

Detection rate of potential human pathogens in SM, BS, and BR samples.
The main potential human pathogens were identified as those with a relatively high median abundance (Fig. 4). In SM samples, those were Streptococcus (10.07%), Corynebacterium (2.14%), Acinetobacter (0.24%), Enterococcus (0.09%), and Pseudomonas (0.03%). In BS samples, these were Mycobacterium (1.31%), Acinetobacter (1.25%), Pseudomonas (0.35%), Aeromonas (0.085%), Bordetella (0.085%), Streptococcus (0.055%), Corynebacterium (0.035%), Bacillus (0.01%), and Enterococcus (0.01%). In BR samples, these were Mycobacterium (0.42%), Bacillus (0.14%), Corynebacterium (0.06%), Pseudomonas (0.06%), Acinetobacter (0.05%), Streptococcus (0.05%), Aeromonas (0.02%), Coxiella (0.02%), and Burkholderia (0.01%).

Relative abundance of potential human pathogens in SM, BS, and BR samples.
Detection rate and median of relative abundance of potential human pathogens were strongly correlated (p < 0.01) based on the Spearman correlation analysis. Potential pathogens satisfying this correlation may pose a huge threat to human health. Using a detection rate of 80% as the threshold, combined with the relative abundance data, we identified Acinetobacter, Corynebacterium, Enterococcus, Pseudomonas, and Streptococcus as major potential human pathogens in SM samples. In BS samples, these are Acinetobacter, Aeromonas, Bordetella, Bacillus, Corynebacterium, Enterococcus, Mycobacterium, Pseudomonas, and Streptococcus. In BR samples, these are Acinetobacter, Bacillus, Corynebacterium, Mycobacterium, and Pseudomonas.
Differences in relative abundance of bacteria and potential human pathogens in SM and digestates
The difference in bacterial relative abundance in SM, BS, and BR and the effect of manure treatment on microbial flora was explored using principal co-ordinates analysis (PCoA) and ANOSIM based on Jaccard distances (Fig. 5). The three sample categories can be well separated (R = 0.63, p = 0.001). ANOSIM confirmed that SM was significantly different from both BS and BR (R = 0.711, p = 0.001 and R = 0.742, p = 0.001, respectively), but the difference was less significant when BS and BR were compared (R = 0.54, p = 0.001). Thus, while fermentation transforms the structure of microbial flora, the latter is similar in BS and residue.

PCoA and ANOSIM analysis of relative abundance of bacteria at genus level in SM, BS, and BR samples. ANOSIM, analysis of similarities; PCoA, principal co-ordinates analysis.
Similar conclusions were obtained after analysis of potential human pathogens after PCoA and ANOSIM analysis (Fig. 6). PCoA shows that the three types of samples can be well separated (R = 0.6, p = 0.001). ANOSIM confirmed the separation of SM from BS and BR (R = 0.644, p = 0.001 and R = 0.76, p = 0.001, respectively), whereas the separation between BS and BR was smaller (R = 0.485, p = 0.001).
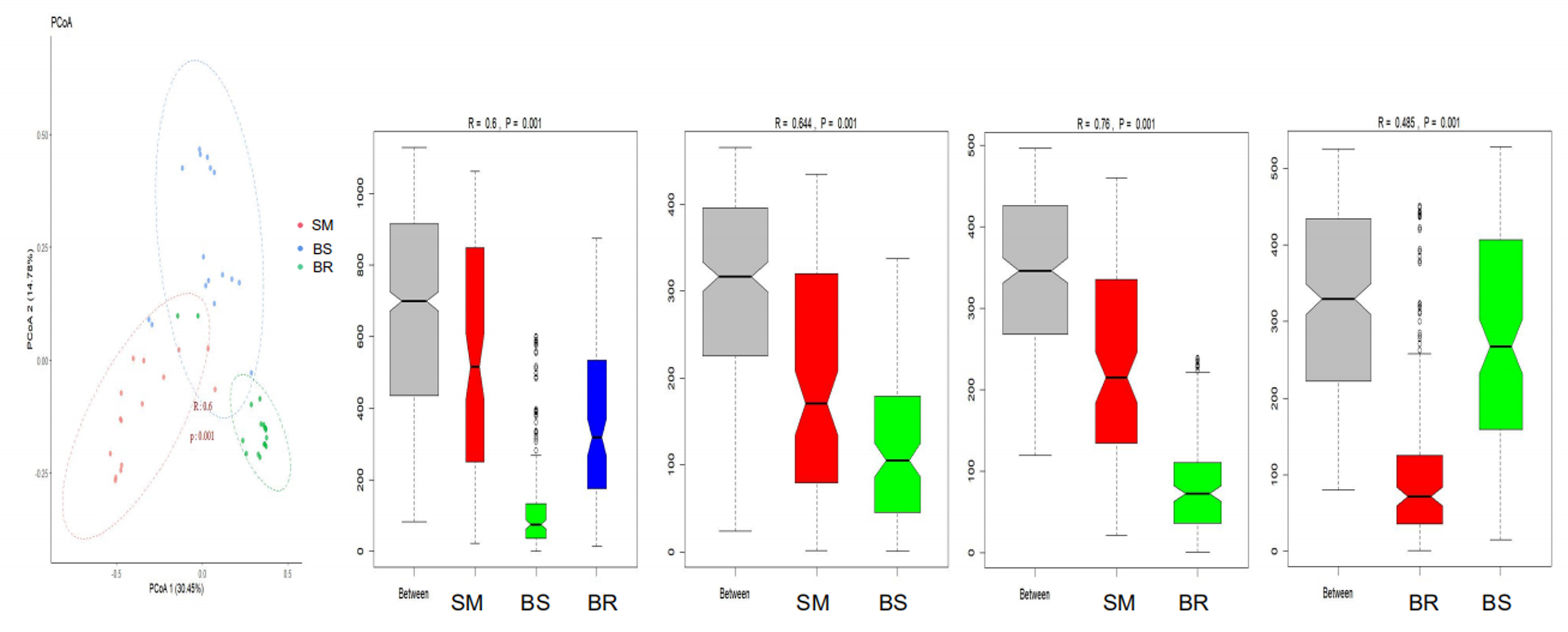
PCoA and ANOSIM analysis of relative abundance of potential human pathogens in SM, BS, and BR samples.
Comparison of major bacteria and human pathogens in SM and digestate
To further understand the impact of fecal treatment process on the abundance of the main bacteria and human pathogens in SM, BS, and BR samples, we applied the Kruskal–Wallis and Bonferroni tests. Using the above results, we selected 11 major bacteria for comparative analysis (Fig. 7). Based on the Kruskal–Wallis test, 11 types of major bacteria were statistically different between the three groups (p < 0.05). Using the Bonferroni test, we compared SM, BS, and BR in pairs.

Comparison of relative abundance of main bacteria in SM, BS, and BR samples. Kruskal-Wallis test results of multi-group comparison were denoted by “”; pairwise comparison results after Kruskal-Wallis test were indicated by “ ”; 0.05 ≥ P > 0.01 marked as *, 0.01 ≥ P > 0.001 marked as **, and P < 0.001 marked as ***.
”; 0.05 ≥ P > 0.01 marked as *, 0.01 ≥ P > 0.001 marked as **, and P < 0.001 marked as ***.
In SM, five kinds of bacteria were statistically more abundant than in BS or BR: Clostridium sensu stricto, Kurthia, Lactobacillus, Streptococcus, and Terrisporobacter. In contrast, fermentation treatment increased the relative abundance of other bacteria: Acidovorax, Flavobacterium, Longilinea, Mycobacterium, Novosphingobium, and Ornatilinea. Thus, while some bacteria are negatively affected by fermentation, others are tolerant to fermentation conditions. However, significant differences were observed in the relative abundance of Acinetobacter, Acidovorax, Clostridium sensu stricto, Flavobacterium, Longilinea, Novosphingobium, and Ornatilinea in BS and BR.
Differences in relative abundance were analyzed selecting nine types of human pathogens (Fig. 8). The Kruskal–Wallis test showed significant differences among SM, BS, and BR (p < 0.01). Analysis of SM, BS, and BR in pairs with the Bonferroni test showed that SM samples had significantly higher relative abundance of Streptococcus, Corynebacterium, and Enterococcus than BS or BR, indicating that these pathogens are controlled by fecal sewage treatment. The relative abundance of Aeromonas Acinetobacter, Bordetella, Bacillus, and Pseudomonas was different in BS and BR.
Comparison of relative abundance of main human pathogens in SM, BS, and BR samples. Kruskal-Wallis test results of multi-group comparison were denoted by “”; pairwise comparison results after Kruskal-Wallis test were indicated by “”; 0.05 ≥ P > 0.01 marked as *, 0.01 ≥ P > 0.001 marked as **, and P < 0.001 marked as ***.
Discussion
Bacteria and pathogens in SM have an important impact on pig's health and are still found in the digestate. The results obtained after combination of detection rate and relative abundance quantification of main bacteria and human pathogens in SM can be interpreted using function information. Clostridium sensu stricto is a beneficial bacterium that produces short-chain fatty acids through anaerobic fermentation that prevent intestinal inflammation (Lopetuso et al., 2013) and can also strengthen the intestinal mucus barrier, inhibiting pathogen adhesion (Wlodarska et al., 2015). Streptococcus suis is known to cause serious porcine disease (Wertheim et al., 2009; Gottschalk et al., 2010) and its presence in SM should be monitored due its potential harm to pigs and humans. Nevertheless, some species of streptococcus are involved in carbohydrate metabolism and reducing the risk of obesity and type 2 diabetes and so on (Jones et al., 2019). Lactobacillus affects the composition of the intestinal flora and regulates the secretion of hormones through the brain/gut axis, reducing human stress and anxiety (Yang et al., 2020). Terrisporobacter, detected abundantly in pig feces, still has not been assigned specific roles.
In this study, we selected five major human pathogens in SM. Corynebacterium is a zoonotic bacterium, mainly related to human or pig diarrhea. Enterococcus faecalis has been reported to affect the cecal microbial community structure in laying hens, improving growth, reducing the diarrhea rate, and increasing beneficial bacteria in weaned piglets (Wang et al., 2019), whereas others have linked it to inflammatory bowel disease, and infective endocarditis have also been reported to be related to Enterococcus faecalis (Escola-Verge et al., 2019; Ruzickova et al., 2020). Streptococcus is abundant in SM, and its presence has to be monitored as discussed above. Acinetobacter spp. are present in fecal, soil, and groundwater ecosystems, with a relative abundance of up to 0.69% of the total microbial community. Acinetobacter is usually considered a symbiotic, opportunistic, relatively low-level pathogen, but it has now been identified in community-acquired infection and constitutes 9% of hospital-infecting pathogens (Pereira et al., 2019). Finally, although Pseudomonas can play a role in inhibiting disease and promoting plant growth, Pseudomonas aeruginosa causes a variety of opportunistic infections, such as eye infections in contact lens wearers, to burns and wound infections, and even lead to septic shock.
In BS, the predominant were Flavobacterium, Novosphingobium, Acidovorax, and Terrisporobacte. Clostridium sensu stricto, Acinetobacter, and Mycobacterium were also present but their relative abundance did not reach 10%. Clostridium sensu stricto, Terrisporobacte, Ornatilinea, and Longilinea were also major bacteria in BR, but their relative abundance was low compared with SM samples. Fermentation provides an environment that may be suitable for the growth of some bacteria. Thus, the detection rate of Flavobacterium, Novosphingobium, Acidovorax, Acinetobacter, and Mycobacterium in BS increased by 50% versus SM. Flavobacterium is a conditional pathogen with a not well-understood pathogenesis. In humans, it causes neonatal meningitis, catheter-associated bacteremia and pneumonia, and is associated with certain cases of HIV disease, therefore, its presence in BS is a potential threat to human health. However, it can degrade polycyclic aromatic hydrocarbons, helping in the control and restoration of soil polycyclic aromatic hydrocarbon pollution. Novosphingobium is a new independent genus, with 49 species reported as on December 2019, related to chronic obstructive pulmonary disease. Its high detection rate in BS may contribute to degrade aromatic compounds, such as 2,4-dichlorophenoxyacetic acid. Acidovorax, a denitrifying bacteria that can convert nitrate nitrogen in BS into nitrogen, can degrade phenanthrene, which is beneficial to control phenanthrene pollution in the soil (Singleton et al., 2018). Mycobacterium includes various pathogenic bacteria such as Mycobacterium tuberculosis, Mycobacterium leprae, and Mycobacterium ulcerans, all with a very high pathogenic potential. However, Mycobacterium can also degrade polycyclic aromatic hydrocarbons and has been isolated from the soil (Zeng et al., 2010).
In BR, the detection rate of Ornatilinea and Longilinea increased to over 50% after fermentation. Longilinea has been linked to be related to the degradation of certain substances, but no function is known for Ornatilinea.
Nine kinds of potential human pathogens were detected in BS and most have been already described above. Aeromonas is a type of organism widely distributed in nature and in agricultural environments and cause a number of infections in humans and animals. Bordetella bronchis is an emerging zoonotic pathogen that can infect. Bordetella bronchis infected a wide range of mammalian hosts. For example, atrophic rhinitis associated with bronchial septicemia has caused a significant loss to the pig industry (Szymczak et al., 2020). Some Bacillus can be used as probiotics for the treatment of intestinal diseases (Liu et al., 2019), whereas Bacillus cereus and Bacillus anthracis are human pathogens. In BR, five major human pathogens were detected, with roles discussed above.
Overall, the composition of bacteria and potential human pathogens in SM, BS, and BR may be potentially harmful both to the environment and to human health. However, although pathogen pollution caused by biogas manure returning to the field should not be underestimated, in some cases it can improve the ability of the soil to degrade harmful substances. Further research is needed to characterize the changes in bacteria and pathogens in SM, BS, and BR to assess the effect of, and to optimize, traditional anaerobic fermentation. Nonparametric comparative analysis can explore further explores the changes in the flora structure and potential human pathogens during SM treatment. This treatment has a significant impact on many major bacteria and human pathogens, but the specific changes taken place should be analyzed independently for each situation.
PCoA and ANOSIM analysis shows that anaerobic fermentation leads to differences in the relative abundance of bacteria and potential human pathogens in SM, BS, and BR. After returning to the field, these population changes may affect the soil microbial environment, and when entering the human body through the food chain they may endanger human health (Bicudo and Goyal, 2003; Hauck et al., 2017).
The Kruskal–Wallis test showed that the absolute abundances of the main bacteria in SM, BS, and BR were different, indicating that anaerobic fermentation has a significant impact in these populations. After fermentation treatment, Mycobacterium increased its relative abundance, but Streptococcus, Enterococcus, and Corynebacterium decreased. Treatment also increased the abundance of some bacteria with special physiological functions, such as Acidovorax and Longilinea. However, even in these cases, the increase of these bacteria in the soil may affect its microecology and the function of soil microorganisms.
Conclusion
Our study has explored the potential harmful effects of SM and its fermentation products to the soil environment and human health. We investigated the structure of the microbial flora in SM, BS, and BR samples of 18 pig farms in the Jiuzhou River of Guangxi, China. Analysis of the structure and abundance of main bacteria and potential human pathogens was based on high-throughput sequencing. Anaerobic fermentation results in higher abundance and diversity of some bacteria, such as Acidovorax, Longilinea, Novosphingobium, and Mycobacterium. The first three have potential material degradation capabilities and may affect the function of the soil itself. However, Mycobacterium is an important human pathogen and may affect human health through insertion in the food chain. Our work is a first step to characterize the structure of bacteria and pathogens in livestock manure and digestate, and provides data to support further for the subsequent research on the harmless treatment of livestock manure and the rational application of biogas fertilizer at the microbiological level.
Footnotes
Acknowledgment
The authors thank EditSprings for editing the English text of a draft of this article.
Author Disclosure Statement
There are no conflicts of interest to declare.
Funding Information
This work was supported by the National Key R&D project (2017YFD0800705), Fundamental Research Fund of Central Public Welfare Research Institutions in 2019 (Innovation team project of new approach and application on substitution toxicology of environmental hormone substance).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.