
Abstract
Klebsiella quasipneumoniae is a recently described species that can be differentiated from Klebsiella pneumoniae. However, in clinical settings, they are frequently misidentified as K. pneumoniae. In this study, our objective was to conduct genomic characterization and bioinformatics analysis of K. quasipneumoniae subsp. quasipneumoniae (KpII-A) isolated from a sample obtained from a retail fish market in Assam, India. Notably, this particular isolate was identified as K. pneumoniae when identified using BD Pheonix™ M50 (BD Difco, USA). This represents a serious pitfall of conventional microbiological methods for distinguishing between K. pneumoniae and K. quasipneumoniae. In this connection, identifying differences in nuclear gene content is key to avoid misidentification. The isolate was confirmed to be KpII-A using species identification by Mash Screen and whole-genome sequencing by the Illumina platform. We report the draft genome sequence of this strain, comprising of 53 contigs with an average GC content of 58.11%. The annotation revealed 5,095 protein coding sequences, 69 tRNA genes, and 4 rRNA genes. The isolated strain acknowledges the presence of oqxA, oqxB, fosA, and blaOKP-A-3 antimicrobial resistance genes (ARGs). Additionally two phage genomes were detected in contigs 3 and 19 of the bacterial genome. Based on the multilocus sequence typing and genome sequencing, the isolate was identified as a novel sequence type, ST5655, within the species K. quasipneumoniae under the phylogroup KpII-A. The presence of antimicrobial resistance genes in KpII-A, isolated from retail fish samples, raises concerns regarding transmission across barriers in ecological niches and possible transmission to consumers. Given that fish may serve as a potential vehicle for ARG transmission, our findings are highly relevant and paramount to human health. Moreover, our study supports the robustness of the sequence-based microbial identification.
Introduction
Klebsiella pneumoniae is a bacterial pathogen that causes hospital or community-acquired infections, which are associated with antimicrobial resistance (AMR) as they possess various AMR determinants. K. pneumoniae established itself as one of the most important causes of global AMR nosocomial infections, especially urinary tract infections, respiratory tract infections, and bloodstream-associated infections (BSIs) (Wyres KL et al., 2019). K. pneumoniae is a Gram-negative, non-motile, non-spore-forming, straight, rod-shaped, and capsuled bacterium. Seven different phylogroups of K. pneumoniae have been identified through genomic analysis: K. pneumoniae sensu stricto (Kp1), K. quasipneumoniae subsp. quasipneumoniae (KpII-A_) (Kp2), K. variicola (Kp3), K. quasipneumoniae subsp. similipneumoniae (Kp4), K. variicola subsp. tropicalensis (Kp5), K. quasivariicola (Kp6), and K. africanensis (Kp7). Within these phylogroups, K. quasipneumoniae isolates, which are closely related to K. pneumoniae, are often misidentified as K. pneumonia (Brisse et al., 2014). These isolates which harbors virulence factors and acquires clinically relevant genes of AMR can cause severe and life-threatening infections similar to the human infections caused by K. pneumonia (S. Wesley Long et al., 2017). K. quasipneumoniae, an emerging pathogenic species and opportunistic bacterium, has the capability to cause AMR infections affecting the gastrointestinal system as well as BSIs in individuals. K. quasipneumoniae has recently been shown to be the causative agent in deadly illnesses. Gram-negative bacteria with extended-spectrum β-lactamase (ESBL) infections are often treated with carbapenems. Clinical isolate strains of K. quasipneumoniae have been shown to exhibit plasmid-mediated carbapenem resistance, which may complicate treatment plans. Since the past 10 years, carbapenemase-producing Enterobacteriaceae has proliferated due to antibiotic selection pressure. OXA-181, carbapenem-hydrolyzing oxacillinase, was initially identified in India in K. pneumoniae and Enterobacter cloacae in 2007. OXA-181 expression in K. quasipneumoniae is rare, and it is predominantly found in K. pneumonia (Min Yi Lau et al., 2020).
Whole-genome sequencing (WGS) is a powerful and a comprehensive method for analyzing the entire genome for identification of bacterial species and for tracking disease outbreaks. The analysis of genomic information through WGS, initially employed in 2013 to investigate outbreaks of foodborne illness. Genome sequencing of multidrug-resistant hypermucoviscous (hm)/hypervirulent (hv) K. quasipneumoniae subsp. similipneumoniae, recovered from the environment in Brazil, was reported for the first time. The findings contribute to a better understanding about the lineages and surveillance studies worldwide (Joao Pedro Rueda Furlan et al., 2020). In this study, we conducted genomic characterization of KpII-A isolated from fish.
Methods
Sample collection and isolation
In March 2019, a KpII-A_strain was isolated from an aquatic ecosystem. The fish sample (Gray Mullet) was collected from retail fish market in Assam, India. The size of the fish sample collected was 0.5–01 kg, which was brought to the laboratory in iced conditions in a thermocol box at 20–25°C. Microbiological analysis was carried out within 3 h of collection. The muscle tissues, including the skin and gut, were homogenized in a sterile Normal Saline solution and 10 mL were transferred in to the Enterobacteriaceae Enrichment Broth (EE Broth Mossel, BD Difco, USA) and incubated at 37°C for 24 h. To isolate ESBL-producing bacteria, a loopful of the enriched culture was inoculated onto MacConkey agar plates (BD Difco, USA) containing 1 g/mL cefotaxime (Sigma-Aldrich, USA). After 24 h at 37°C, based on the morphological characteristic of Klebsiella sp., large pink mucoid colonies were chosen. These colonies underwent additional purification, i.e., the colonies selected were grown overnight on tryptic soya agar and eosin methylene blue agar for DNA extraction (TSA, EMB, BD Difco, USA). Antibiotic susceptibility was identified and evaluated using the BD PhoenixTM M50 automated system (BD Difco, USA). The results were interpreted in accordance with Clinical and Laboratory Standards Institute recommendations (CLSI, 2020). Following the manufacturer’s instructions, genomic DNA was isolated using the MasterPureTM DNA purification kit from (Lucigen in California). In this study K. quasipneumoniae subsp. quasipneumoniae strain isolated is referred as ‘KpII-A_’.
Whole-genome sequencing and analysis
MicrobesNG, Birmingham, UK, employed the Illumina HiSeq2500 sequencing platform for the genomic sequencing. A paired end library was created using Illumina, CA, USA), and the entire genome’s quality was checked using FastQC (Andrews, 2010). After that, the genome was assembled using Shovill v. 1.0.4 (available at https://github.com/tseemann/shovill), a bioinformatics tool specifically designed for bacterial genome assembly from Illumina paired-end sequencing data. Shovill integrates several well-established software tools and pipelines, such as SPAdes and BWA, to streamline the assembly process. Mash Screen v. 2.3 (Ondov et al., 2019) and ribosomal multi-locus sequence typing (rMLST) (https://pubmlst.org/species-id/) were used to test the assembled genome for species identification. Average nucleotide identity (ANI) was performed between novel K. quasipneumoniae (KpII-A_) and the reference genome G4584 (K. quasipuemoniae, RefSeq accession no. NZ_CP034129 NZ_NJCN01000000 NZ_NJCN01000001-NZ_NJCN01000134) which involves genome sequence fragmentation, nucleotide sequence search, alignment and identity calculation using Basic Local Alignment Search Tool (BLAST) program as its search engine. ANI with more than 95% identity was considered suitable to identify species and ≥98% to identify subspecies. Assembled genome was submitted to comprehensive genome analysis service at PATRIC-assisted RASTtk-enabled genome annotation pipeline (Brettin et al., 2015; Davis et al., 2020). Downstream analysis of the genome was conducted by adopting various online bioinformatics platforms. Antibiotic resistant genes (ARGs) were identified using ResFinder 4.1 (https://cge.cbs.dtu.dk/services/ResFinder/) with default parameters (≥80% identity over ≥60% of the length of the target gene). Comprehensive antimicrobial resistance database, CARD (https://card.mcmaster.ca/analyze/rgi/) is also used to confirm the AMR genes using a sequence analytical tool. Resistant Gene Identifier (RGI) which accepts the WGS assembly contigs (maximum size, 20 Mb) provides the AMR genes and targeted drug classes (Andrew G. McArthu et al., 2013). Presence of virulence factors were screened with VFanalyzer (http://www.mgc.ac.cn/VFs/). Plasmid replicon was identified using PlasmidFinder 2.1 (https://cge.cbs.dtu.dk/services/PlasmidFinder/) in which the percentage identity (%ID) threshold must be at least 80% nucleotide identity with the currently included replicon sequence in the database. The K and O antigens were determined using Kaptive v. 0.7.3 (http://kaptive.holtlab.net/) (Wick et al., 2018). The whole-genome was used to generate the MLST of the isolate in the web portal (https://pubmlst.org/bigsdb?db=pubmlst_mlst_seqdef&page/). Phage genomes were identified by using online tool PHASTER (PHAge Search Tool Enhanced Release) (https://phaster.ca/) in which ∼90% of the prophages can be identified in contigs ≥20,000 bp long and PHASTER’s runtime is ∼210 s for a raw genome sequence and ∼100 s for a pre-annotated GenBank input (David Arndt et al., 2016). A genome comparison of the draft genome ‘KpII-A_ was performed using BLAST Ring Image Generator (BRIG) in which all BLAST comparisons and file parsing were done automatically via a simple GUI, and the images show similarity between a central reference sequence and other sequences as concentric rings.
Results
The assembled genome consisted of 53 contigs, totaling 5,256,469 bp in length, with an average G ± C content of 58.11%. The annotation process revealed the presence of 632 hypothetical proteins and 4,486 proteins with functional assignments. The N50 length, representing the shortest set of sequence length at 50% of the reported assembly length, was determined to be 329,458 bp. Additionally, the L50 count, which signifies the smallest number of contigs needed to produce the N50 length, was found to be 5. Among the proteins with functional assignments, 1,438 were associated with Enzyme Commission (EC) numbers, 1,191 had Gene Ontology (GO) assignments, and 1,064 proteins were mapped to KEGG pathways. The annotation process conducted by PATRIC included the classification of proteins into two categories: 4,932 proteins belonged to genus-specific protein families (PLFams), while 4,942 proteins were categorized under cross-genus protein families (PGFams). This particular genome was found to possess a total of 5,095 protein coding sequences, along with 69 transfer RNA (tRNA) genes and 4 ribosomal RNA (rRNA) genes.
The whole-genome sequencing analysis of the query genome ‘KpII-A_’ was confirmed as K. quasipneumoniea using KmerFinder3.2 and was assigned to the phylogroup Kp2. ANI, a web-based tool. The tool was utilized to determine the average nucleotide identity with both best hits One-way ANI 1: 96.27% from 22648 fragments and reciprocal best hits two-way ANI: 96.40% from 21492 fragments. The analysis was carried out between two genomic datasets, ‘KpII-A_’ K. quasipuemoniae strain and the reference genome G4584 (K. quasipuemoniae, RefSeq accession no. NZ_CP034129 NZ_NJCN01000000 NZ_NJCN01000001-NZ_NJCN01000134), which is shown in Figure 1. Multilocus sequence typing (MLST) classification using seven housekeeping genes gapA, infB, mdh, pgi, phoE, rpoB, and tonB reveals a new MLST designation, ST5655. The sequence type 5655 was assigned to the strain with the allelic profile such as gapA_17, infB_19, mdh_39, pgi_39, phoE_315, rpoB_21, and tonB_126.
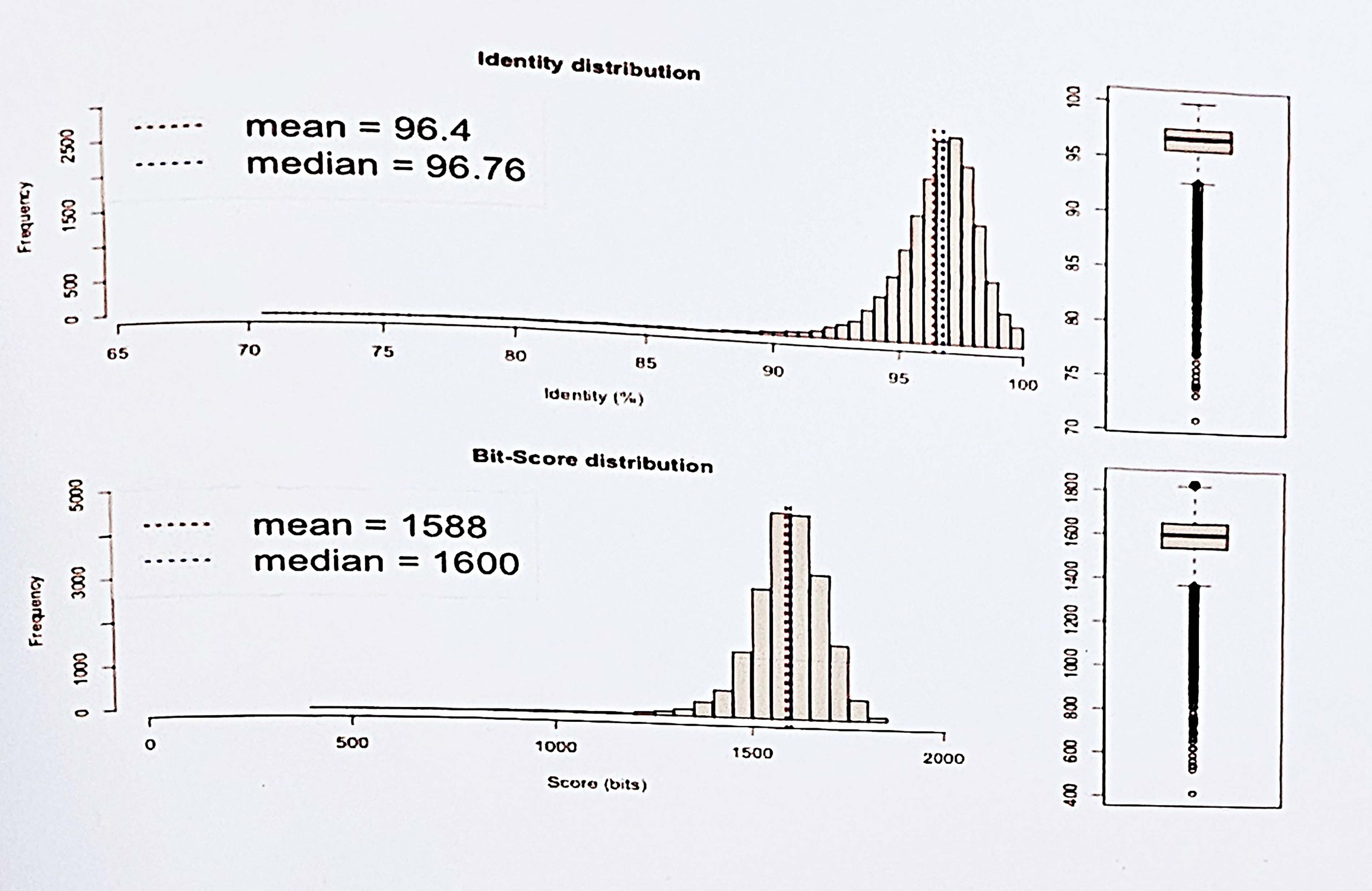
Estimation of Average Nucleotide Identity between the genome KPII-A_ and the reference genome strain G4584 using ANI (Average Nucleotide Identity) calculator using both best hits and reciprocal best hits between the two genomes. The bit score represents the statistical measure of sequence similarity between two genomic regions. The comparison shows an average ANI of 96.4%.
Antimicrobial resistance pattern
The antimicrobial resistant (AMR) gene in KpII-A_ includes oqxA and oqxB genes, which are resistant to trimethoprim, ciprofloxacin, cetylpyridinium chloride, nalidixic acid, benzylkonium chloride, and chloramphenicol; fosA and fosA6 genes, which are resistant to fosfomycin; and blaOKP-A-3 gene, which is resistant to unknown beta-lactam. WGS-based antimicrobial susceptibility testing (AST) shows no resistance to ampicillin, temocillin, cephalothin, cmoxicillin±clavulanic acid, cefoxitin, and cefotaxime antibiotics. A K-mer-based AMR genes detection method in PATRIC with specific AMR mechanisms is provided in Table 1. Plasmid Cl440I with a 95.61% identity was recorded. Two prophage regions have been identified, of which 1 region is intact in contig-3 with a Guanine and cytocine (GC)% of 50.47% and 1 region is incomplete in contig-19 with a GC% of 53.99% and other regions are questionable. PHAGE_Salmon_118970_sal3_NC_031940 with a region length of 38.5 Kb is identified in contig-3 of KpII-A_ genome. The functional phage profile of the prophage is represented in Figure 2. The comparative genomic study reveals the highest similarity between the query genome ‘KPII-A’ and multiple reference genomes, as shown in Figure 3 using the software BRIG. The reference genomes of K. quasipneumoniae species used in this comparative genomic study were extracted from the NCBI database and the strains selected are from clinical and environmental samples. Additionally, in the present study, the phylogenetic tree of the draft genome KpII-A_ and other K. pneumoniae species from the NCBI database was generated using PATRIC, as represented in Figure 4.

Linear genomic map of one intact prophage sequence

Simulated BRIG output image shows a comparison of the draft genome K. quasipneumoniae (KPII-A) against five reference genomes (WW-14A, G4584, GDQ8D117M, KqPF26, KW1) with an accession number NZ_CP080099, NZ_CP034129, NZ_MK618661, NZ_CP065838, NZ_CP102898. The innermost rings show GC content (black) and GC skew (purple/green). The draft genome of KpII-A has 70–100% identity with all the five reference genomes.
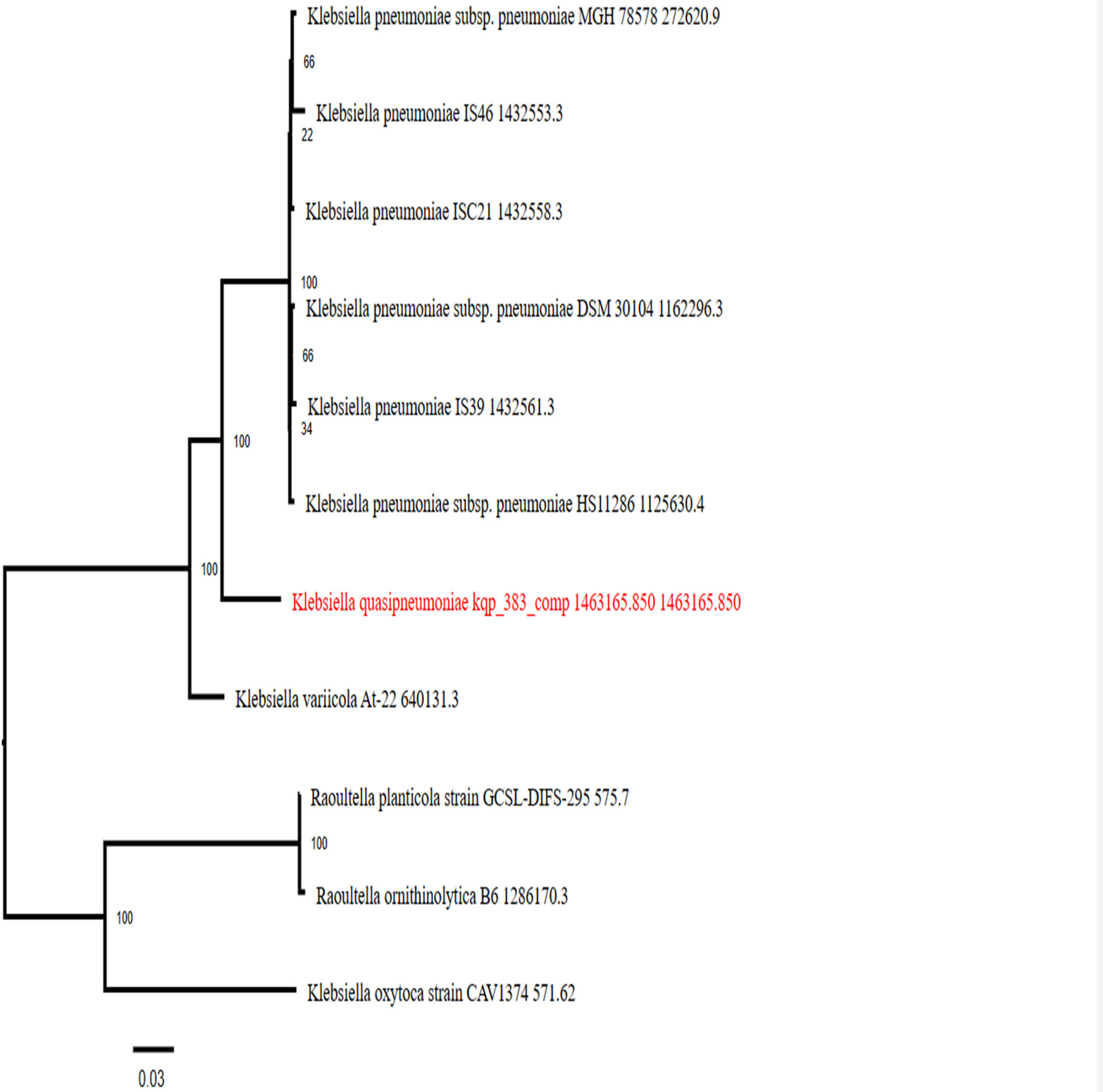
PATRIC provides phylogenetic tree of draft genome KpII-A shows the relationship between the draft genome and other Klebsiella species. Bootstrap method is used to generate the supporting values in the tree.
AMR-Related Genes with Specific AMR Mechanisms

K-mer-based antimicrobial resistance genes of K. quasipneuoniae (KpII-A) with the specific AMR mechanisms using the PATRIC’s curated collection of representative AMR gene sequence variants.
AMR, antimicrobial resistance.
Discussion
K. pneumoniae is one of the most common cause of deadly infections. K. quasipneumoniae was previously thought to be an opportunistic pathogen with lower virulence in humans compared to K. pneumoniae. Nevertheless ESBL-producing K. quasipneumoniae and ESBL-producing K. pneumoniae strains are similar in their virulence which results in intrusive infections and death rates that are analytically comparable to those of K. pneumoniae strains. K. quasipneumoniae are prominent in gastrointestinal tracts of animals and humans when the rare opportunistic pathogens are consumed through food. Although K. quasipneumoniae genome is different from K. pneumoniae, they share similarity in the virulence genes, MLST and plasmid-mediated antibiotic resistance which leads to the mistake in the interpretation of species. Based on this misconception a study is reported, the misidentification of K. quasipneumoniae as K. pneumoniae (Long et al., 2017). The limited capability of traditional clinical microbiology laboratory methods to differentiate K. quasipneumoniae from K. pneumoniae could lead to an underestimation of their capacity to cause severe infections in humans. WGS, a standardized approach for an easier and accurate identification and diagnose.
Gray mullets, Mugil cephalus are one of the most economically important cultivable species alongside other mullet species. As per reports, the farming of gray mullet, has been taking place in Kerala, West Bengal, and Tamil Nadu since the 1940s. Contemporary aquaculture practices involve the use of inorganic chemicals and fertilizers to treat ponds. Farmers are also using artificial feeds and overstocking ponds to promote accelerated growth of fish. However, these modern methods can result in stress on the farmed fish, which may increase their susceptibility to infections caused by various types of pathogenic bacteria (Mastan, 2013). Among the known pathogenic bacteria, Klebsiella species are found to be infectious to various organisms including fish. Continuing AMR surveillance is crucial in all sectors, including fisheries, in this regard. In our study, the WGS of the isolate (KpII-Ap) from the fish sample (gray mullet) demonstrate the genetic context of KpII-A. Though there are various β-lactamase genes among Enterobacteriaceae blaOKP, a known chromosomal species-specific marker for K. quasipneumoniae, was found to be uniquely restricted within the K. quasipneumoniae cluster. The precise identification of blaOKP variant can be critical for the epidemiological purpose and infection control. The blaOKP genes are known to have numerous variants that are divided into the two major subgroups blaOKP-A and blaOKP-B (Fevre et al., 2005; Long et al., 2017; Nicolás et al., 2018). FosA gene is a glutathione transferase, often found in the chromosome of K. pneumonia which inactivates fosfomycin through catalyzing the addition of glutathione The presence of fosA6 plasmid-encoded enzyme confers resistance to fosfomycin. The oqxAB efflux pump, which are encoded by two genes oqxA and oqxB are prevalent in Enterobacteriaceae over the last decade and has become a crucial area of study (Kulková N et al., 2014). Expression of oqxAB efflux pump genes reduces susceptibility to classes quinolones, amphenicole, folate pathway antagonist and quaternary ammonium compound (Rodríguez-Martínez et al., 2013). K. quasipneumoniae (KpII-A) harbors plasmid Col440I. The Col440I plasmid type was found to be highly conserved by displaying qnrB19, a pspF operon, and various genes with unknown functions (Katharina Juraschek et al., 2022). The percentage of GC content and the length of the genome are similar to that of the reference genome. The N50 value is used to interpret the average length of the contigs, and in the current thesis’s N50 is lower which can be caused by the production of large number of contigs that results in shorter contigs. The total length of the contigs is consistent when compare to the results of a previous study on K. pneumoniae (Runcharoen et al., 2017). In addition, it is possible to assert that all isolates, including those that were mistakenly identified as K. pneumoniae, had genome fractions that were relatively low. The fact that the software for assembly in the previous study was different from the one used in this thesis may also account for the difference in results between the two studies.
In our study, the analysis to find the presence and absence of AMR genes was carried out using the ResFinder, the tool that provides phenotypic antibiotic resistant genes with an identity of about 99.74%. The tool recognizes AMR genes using the fasta files assembled by SPAdes (Zankari et al., 2012). As the identification of the species, K. quasipneumoniae and K. variicola has been mistaken as K. pneumoniae (Berry et al., 2015; Seki et al., 2013) in different studies and reports because of the similarity and the difficulty in differentiating among the Klebsiella phylogroups. The genotypic identification of species helps for an accurate interpretation of the species (Long et al., 2017). The genotypic identification of species is carried out by the Center for Genomic Epidemiology (CGE) in which the data from Kraken, PathogenFinder, ResFinder and “species” are used. SpeciesFinder in CGE tool was not able to differentiate several Klebsiella isolates from other bacteria such as many of the isolates of K. oxytoca and K. quasipneumoniae had an unknown species. An alternative method was used, KmerFinder to obtain the results and recognize the species, a batch was uploaded in the CGE website and executed, the alternative method recognizes a majority of isolates of K. pneumoniae and K. variicola are recognized. The blaLEN type, which is shared by half of the K. pneumoniae and K. variicola isolates, was found to be expressed when the ResFinder result was examined. It was also noted that the blaOKP type of beta-lactamase in several K. pneumoniae isolates acts as biological marker to differentiate between the K. pneumoniae phylogroup. Earlier, this identification was made when isolates were misidentified and assumed to be K. pneumoniae which were reclassified as K. quasipneumoniae by the expression of the OKP-type beta-lactamase (Becker et al., 2018; Long et al., 2017; Shankar et al., 2017). Similar to the present findings, Sudha et al. (2022) reported the genomic characterization of K. quasipneumoniae subsp. similipneumoniae (India238 strain) ST1699 and serotypes KL52 and OL103 isolated from fish.
The phenotypic antibiotic resistance confirmed through the AST in the laboratory may be different from that of the results obtained from the analysis part. The main reason behind this inconsistency is the fewer cases of resistance observed in phenotypic AST compared to the predictions made by ResFinder. However, a significant limitation of genotypic methods is that the detected genetic markers may not always result in expression and translation of phenotypic resistance. Consequently, false resistance can be anticipated for genetic markers that are tightly regulated, including efflux pumps or inducible β-lactamases (Ferreira et al., 2020). The results of various studies on Klebsiella species were contrasted with the results of our study which shows an interesting fact that strain of K. pneumoniae, K. variicola, K. quasipneumoniae, and K. quasivariicolao that shows resistance to fluoroquinolones also contain oqxAB gene. The prophage in the host genome have an impact on their genome which can cause change in their level of virulence or antibiotic resistance. The abundance and diversity of prophage genomes in prokaryotes, particularly those belonging to the Enterobacteriaceae family, had been highlighted in one recent study. In fast-growing bacteria and bacteria with large genome, the prophage occurs more frequently. In the present study only one intact and one incomplete prophage regions were present in which the intact region under goes a selection pressure and genetic degradation. The remaining prophages were classed as questionable.
Conclusion
The present study of KpII-A contributes to a better understanding of its phenotypic and genotypic characteristics. Moreover, our study supports the robustness of sequence-based microbial identification. The bioinformatics approach for the interpretation of the actual gene products provides more nationally relevant data. The presence of AMR genes in KpII-A isolated from retail fish samples raises concerns related to transmission across barriers in ecological niches and possible transmission to consumers. Given that fish may serve as a potential vehicle for transmission of ARGs, our findings are of paramount importance for human health. The AMR genes nurturing K. quasipneumoniae, which has been isolated from the fish samples requires attention and the food sources must be under scrutiny. The study of the molecular epidemiology of AMR pathogens, their genetic association, and virulence genes helps to control the spread of zoonotic diseases. The ability of beta-lactamase-producing bacteria to spread AMR genes have a considerable impact on the environment. The multi-drug resistant K. quasipneumoniae with an ST-5655 and a phage genome (PHAGE_Salmon_118970_sal3_NC_031940) isolated from the fish sample poses threat to the human health.
Genome Accession
The genome sequences in this study have been deposited in NCBI under accession number JASBCT00000000. Genome database under BioProject PRJNA965951 and Biosample SAMN34510381.
Authors’ Contributions
Conceptualisation and design of the study: G.K.S., B.R.S., and M.A.H. Fund acquisition: G.K.S., B.R.S., and M.A.H. Data acquisition and result interpretation: G.K.S., Su.S., and M.K.H. DNA sequence and edited: C.R., M.K.H., and Sa.S. Draft article preparation: Sa.S. and G.K.S. Review and editing of article: G.K.S. and M.A.H. All the authors critically reviewed and approved the article for the publication.
Footnotes
Author Disclosure Statement
Each of the authors declares that the manuscript has not been previously published and is not currently under the consideration by any journals. All the authors have critically evaluated and approved the contents. Each named author has significantly contributed to conducting the underlying research and drafting the manuscript and declares no conflict of interests financially or otherwise.
Funding Information
This study was supported by the Department of Biotechnology, Government of India (BT/IN/Indo-UK/AMR/06/BRS/2018–19), and Economic and Social Research Council, UK.