
Abstract
Conobacter sakazakii represents a significant foodborne pathogen threatening susceptible populations, yet its molecular characteristics in powdered spices and instant cereals remain poorly characterized. This investigation analyzed 45 retail samples (20 spices, 25 cereals) collected from Nanning, China, through pathogen isolation, whole genome sequencing, and bioinformatics profiling to elucidate virulence determinants, antimicrobial resistance patterns, and genetic features. The study revealed an overall contamination rate of 22.2% (10/45), with contamination rates of 25.0% in spices and 20.0% in cereals. Predominant serotype O1 accounted for 70.0% of isolates, while multilocus sequence typing identified seven sequence types, including novel ST1008. Genomic analysis demonstrated high genetic diversity (allelic differences: 57–2678) and revealed 42 virulence-associated genes (including flgA, ompA, and rpoS) alongside multidrug resistance patterns (notably to selected β-lactams). Crucially, mobile genetic element-mediated mcr-9 (conferring colistin resistance) and astA (encoding heat-stable enterotoxin) were detected, indicating significant transmission risks. This study provides the first documentation of concurrent virulence-resistance characteristics in C. sakazakii from Chinese retail powdered foods, highlighting the need for enhanced surveillance throughout low-moisture food supply chains to mitigate infection risks in vulnerable populations.
Introduction
Cronobacter species (spp.) are emerging foodborne pathogens comprising seven distinct species, with Cronobacter sakazakii (C. sakazakii) being the most clinically significant (Iversen et al., 2008; Joseph et al., 2012). C. sakazakii, a facultative anaerobic Gram-negative bacillus, colonizes human and animal intestinal tracts and poses a particular threat to infants, young children, and immunocompromised individuals. The pathogen demonstrates remarkable environmental resilience, surviving under acidic conditions (pH <3.9), elevated salt concentrations (10% NaCl), and desiccation. It has been isolated from various food sources, including infant formula, dietary supplements, processed meats, dairy products, and spices (Arslan and Ertürk, 2021).
Global surveillance data indicate that the annual incidence of C. sakazakii infection ranges from 1 to 3 cases per 100,000 infants, with mortality rates reaching 40–80% in severe cases (Bowen and Braden, 2006; Strysko et al., 2020). While much of the existing research has focused on C. sakazakii infections in infants, particularly those associated with powdered infant formula, recent evidence suggests that this pathogen also poses a significant threat to adult health. In fact, adults are affected more frequently than infants, with clinical manifestations including bacteremia, urinary tract infections, diarrhea, and acute cholangitis (Hayashi et al., 2021; Lepuschitz et al., 2019; Sahra et al., 2021). Furthermore, C. sakazakii has been identified as a key agent in foodborne acute gastroenteritis outbreaks among healthy adults (Yong et al., 2018). Despite these findings, there remains a paucity of research on the molecular characteristics, virulence potential, and pathogenicity of C. sakazakii in foods consumed by adults.
Given China’s pivotal role in global food production, enhanced surveillance of C. sakazakii contamination is imperative. The pathogen’s virulence is mediated by multiple factors, including adhesins, iron acquisition systems, and outer membrane components (Mohammadgholi Pour et al., 2024). Of particular concern is its demonstrated resistance to a broad spectrum of antimicrobial agents, driven by various resistance genes and mobile genetic elements (MGEs) (Shi et al., 2018).
This study focuses on powdered spices and instant cereals due to their widespread consumption across all age groups, including high-risk populations such as infants, young children, and immunocompromised individuals. Spices are commonly used in daily cooking and are consumed by the general population, while instant cereals are popular among young children, middle-aged, and elderly individuals in China due to their convenience and nutritional value. These food matrices are particularly susceptible to contamination due to their low-moisture content and processing conditions, which align with C. sakazakii’s known tolerance to desiccation. Therefore, retail samples of powdered spices and instant cereals were collected in Nanning City, China, and C. sakazakii strains isolated from these samples were analyzed using whole genome sequencing (WGS) to investigate their genomic features and pathogenic potential.
The primary aim of this study is to characterize the genomic diversity, evolutionary relationships, and variations in antimicrobial resistance and virulence determinants of C. sakazakii strains isolated from powdered spices and instant cereals. These findings will advance our understanding of the molecular characteristics and pathogenic potential of C. sakazakii in these widely consumed food products, thereby informing the development of improved food safety measures and public health strategies.
Materials and Methods
Sample collection
In this study, a total of 45 retail samples, comprising 20 raw powdered spices and 25 instant cereals, were collected from the retail markets and supermarkets in Nanning City, China, in 2023. All samples were tested for C. sakazakii contamination following the national standard method GB 4789.40-2016 (currently updated to GB 4789.40-2024, National Health Commission of China and State Administration for Market Regulation, 2024). Detailed information on the time and locations of the sample collection is provided in Supplementary Table S1.
Bacterial isolation and identification
Each 100, 10, and 1 g sample portion was mixed with 900, 90, and 9 mL of sterile buffered peptone water, respectively, at a 1:9 ratio (weight to volume). This process was repeated in triplicate. After incubation at 36°C for 18 h, 1 mL of each enrichment culture was transferred to 10 mL modified lauryl sulfate tryptose broth-vancomycin (mLST-Vm; HKM, Guangzhou, China) and incubated at 44°C for 24 h. The cultures were then streaked onto Enterobacter sakazakii chromogenic agar (CHROMagar, Paris, France), where blue-green colonies were considered presumptive C. sakazakii. Suspected colonies were further isolated on tryptic soy agar (TSA; HKM, Guangzhou, China), and yellow-pigmented colonies were selected for protein extraction using the formic acid method. A 1 μL aliquot of the supernatant was spotted onto a target plate, air-dried, and then overlaid with 1 μL of α-cyano-4-hydroxy-cinnamic acid matrix. After air-drying, the samples were analyzed using matrix-assisted laser desorption/Ionization time of flight mass spectrometry (MALDI-TOF-MS; Bruker, Billerica, MA, USA) in accordance with the national standard method. The mass spectra were recorded and compared against the Bruker MALDI Biotyper reference database for species identification.
Serotyping by qPCR
Bacterial isolates identified as C. sakazakii by MALDI-TOF-MS were further characterized by serotyping. Genomic DNA was extracted using a commercial DNA isolation kit (Qiagen, Hilden, Germany). Serotype identification was performed using quantitative real-time fluorescence Polymerase Chain Reaction (qPCR; ABI7500, Thermo Fisher Scientific, MA, USA) targeting the wzy and wzx genes, as previously described (Sun et al., 2012). Seven pairs of primers specific to the wzy and wzx genes were synthesized for this purpose. The PCR amplification protocol included an initial denaturation at 95°C for 3 min, followed by 40 cycles of denaturation at 95°C for 5 s and annealing at 60°C for 31 s. A melting curve analysis was subsequently performed with the following steps: 95°C for 15 s, 60°C for 1 minute, 95°C for 15 s, and 60°C for 15 s. The complete primer list is provided in Supplementary Table S2.
Whole genome sequencing
Genomic DNA was extracted from confirmed C. sakazakii isolates using a bacterial DNA extraction kit (Qiagen, Hilden, Germany). WGS was performed on a Genetic Sequencer platform (MGI, Shenzhen, China), achieving a minimum coverage depth of 80-fold. Low-quality reads were filtered during data preprocessing, and the cleaned sequence data met the required quantity and quality thresholds. The filtered reads were assembled using SPAdes v3.13.0, yielding an average assembly N50 value exceeds that 150 kb (Supplementary Table S3).
Sequence typing of C. sakazakii isolates
The PubMLST database (https://pubmlst.org/), which contained 4894 Cronobacter species records, and 1215 C. sakazakii-specific entries as of November 2024 were utilized for sequence typing. Seven housekeeping gene sequences (atpD, fusA, glnS, gltB, gyrB, infB, pps) were extracted and queried against the Cronobacter MLST database for conventional MLST typing. Additionally, a core-genome MLST (cgMLST) analysis was performed using a 2,831-target scheme developed for C. sakazakii. Minimum spanning trees were constructed using PHYLOViZ Online (https://online2.phyloviz.net/index) to establish genotypic relationships among the isolates. Phylogenetic analysis was conducted using the neighbor-joining method and Tamura-Nei model in MEGA 7.0 software, and the results were visualized with the iTOL online tool (https://itol.embl.de/) (Letunic and Bork, 2024). Sequence comparisons and nucleotide similarity analyses were performed using EMBL-EBI tools (Madeira et al., 2024). Finally, the assembled genomes were annotated using the Prokka software, and circular chromosomes were visualized with the CGView tool (https://cgview.ca/).
Detection of virulence genes and antibiotic resistance
The assembled genomes were annotated using the Prokka software to identify virulence genes associated with C. sakazakii pathogenicity. The distribution and variations of these genes were further characterized using the comprehensive antibiotic resistance database to predict resistance mechanisms and transmission patterns (Alcock et al., 2023).
Antibiotic susceptibility
Antibiotic susceptibility was assessed using an Automated Microbiology System (BD, NYC, USA). Commercial reagent kits were employed, covering the following antibiotics: amikacin, amoxicillin-clavulanic acid, ampicillin-sulbactam, aztreonam, colistin, ertapenem, nitrofurantoin, ciprofloxacin, trimethoprim-sulfamethoxazole, chloramphenicol, meropenem, minocycline, moxifloxacin, norfloxacin, piperacillin-tazobactam, gentamicin, tetracycline, tigecycline, cefepime, cefuroxime, ceftriaxone, ceftazidime, cefoxitin, cefazolin, tobramycin, imipenem, and levofloxacin. Resistance and susceptibility profiles were determined in accordance with the manufacturer’s instructions and Clinical and Laboratory Standards Institute guidelines. Escherichia coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853 were used as quality control strains.
Detection of plasmids and mobile genetic elements
Plasmids and MGEs were detected using the tools available from the Center for Genomic Epidemiology (http://www.genomicepidemiology.org/). The identity thresholds for plasmid and MGE identification were set at 92.17% and 90.54%, respectively.
Results and Discussion
Prevalence of C. sakazakii in powdered spices and instant cereals in Nanning, China
Ten C. sakazakii strains were isolated from 45 food samples in Nanning, China, with contamination rates of 25.0% (5/20) in powdered spices and 20.0% (5/25) in instant cereals (Supplementary Table S1, S4). These rates are notably higher than those reported in previous studies on dried edible mushrooms (14.8%) (Jiang et al., 2022), aquatic products (3.90%) (Li et al., 2020), and meat products (9.18%) (Zeng et al., 2020). The elevated contamination levels in powdered spices and instant cereals suggest that these food types may serve as potential reservoirs for C. sakazakii. The higher prevalence may be attributed to their low-moisture content and specific processing conditions, such as drying and dehydration, which align with C. sakazakii’s known tolerance to desiccation (Mohammadgholi Pour et al., 2024).
Genetic diversity and epidemiological relevance of serotypes and sequence types
Serotyping revealed O1 as the predominant serotype (70.0%, 7/10), followed by O2 (20.0%, 2/10) and O4 (10.0%, 1/10) (Table 1). Genetic analysis revealed substantial diversity, with the number of allelic differences ranging from 57 to 2,678 across isolates. Notably, ST8 (strain C8) and ST226 (strain C10) from clonal complex 8 (CC8) exhibited 849 allelic differences, while other sequence types showed over 2630 allelic variations (Fig. 1). Seven distinct STs were identified, including a novel ST designated as ST1008, with consistent serotypes among isolates of identical sequence types. Predominant STs included ST1, ST17, and ST226 (20.0% each), while ST8, ST40, ST58, and ST1008 each accounted for 10.0%.
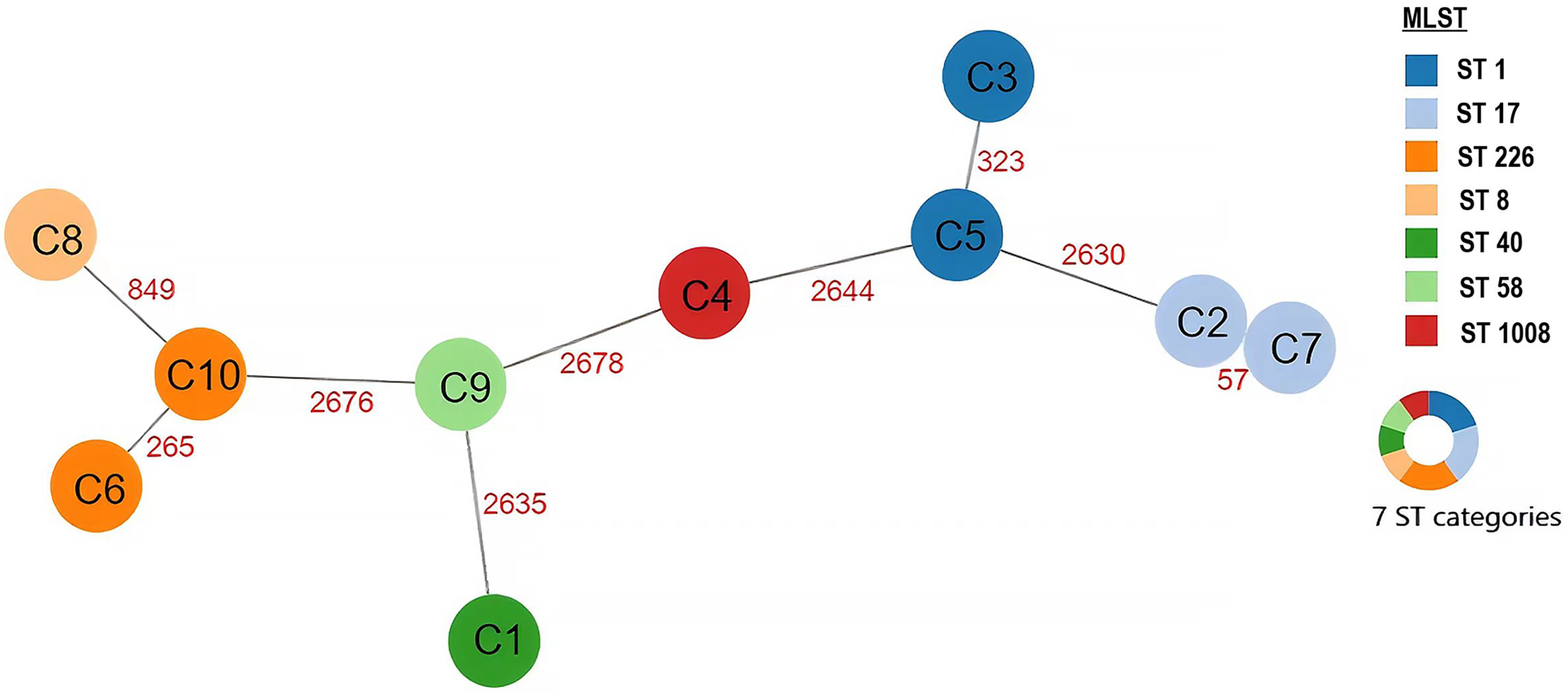
The minimum spanning tree (MST) of 10 Cronobacter sakazakii strains. Phylogenetic analysis was performed using PHYLOViZ Online (https://online2.phyloviz.net/). Sequence types (STs) are represented by differently colored circles. Red numbers on connecting lines indicate the number of allelic differences between isolates based on the core-genome multilocus sequence typing (cgMLST) scheme, which comprised 2831 gene loci for C. sakazakii.
MLST Typing of 10 Strains of Cronobacter sakazakii
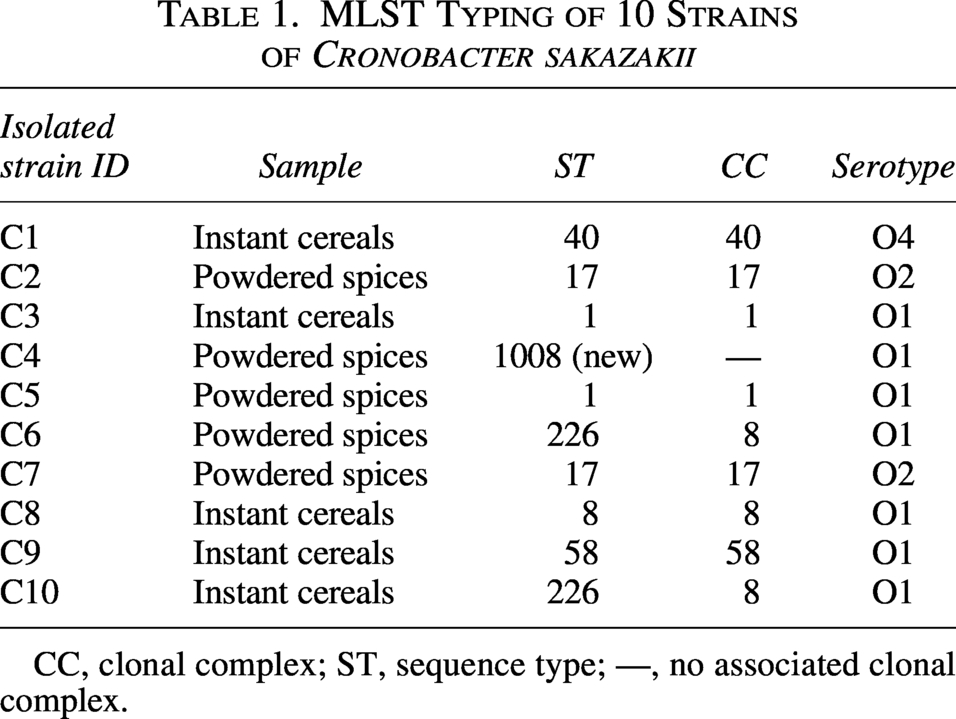
CC, clonal complex; ST, sequence type; —, no associated clonal complex.
Housekeeping gene analysis (atpD, fusA, glnS, gltB, gyrB, infB, and pps) revealed distinct genetic relationships (Fig. 2). For example: Strains C3 and C5 (ST1) clustered within clonal complex 1 (CC1) alongside ST693 and ST769. Strains C2 and C7 (ST17) grouped with ST1007 and ST825 in CC17. The novel ST1008 (C4) exhibited 99.24–99.34% concordance with ST823/ST630 but showed 2644 allelic variations via cgMLST, indicating a unique evolutionary lineage (Figs. 2 and 3). The circular genome of the ST1008 strain is illustrated in Figure 3.
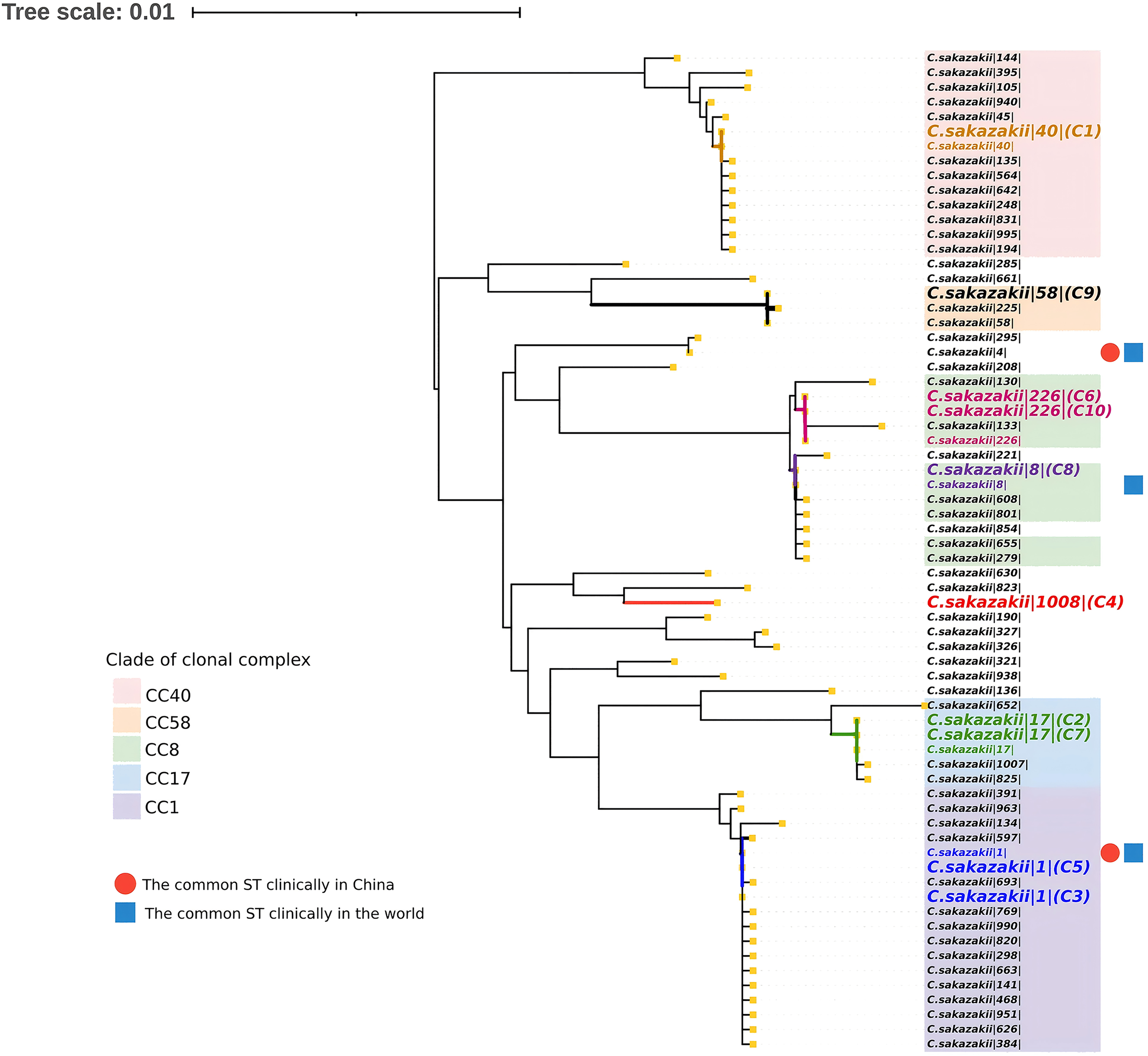
Phylogenetic analysis of 10 Cronobacter sakazakii strains. Neighbor-joining (NJ) tree constructed using seven housekeeping gene loci with MEGA 7.0. Clonal complexes are distinguished by color-coded branches. Metadata for each sequence type (ST) is presented on the right. Orange squares indicate isolated origins. Red dots mark clinically predominant STs in China, while blue squares highlight globally predominant STs.
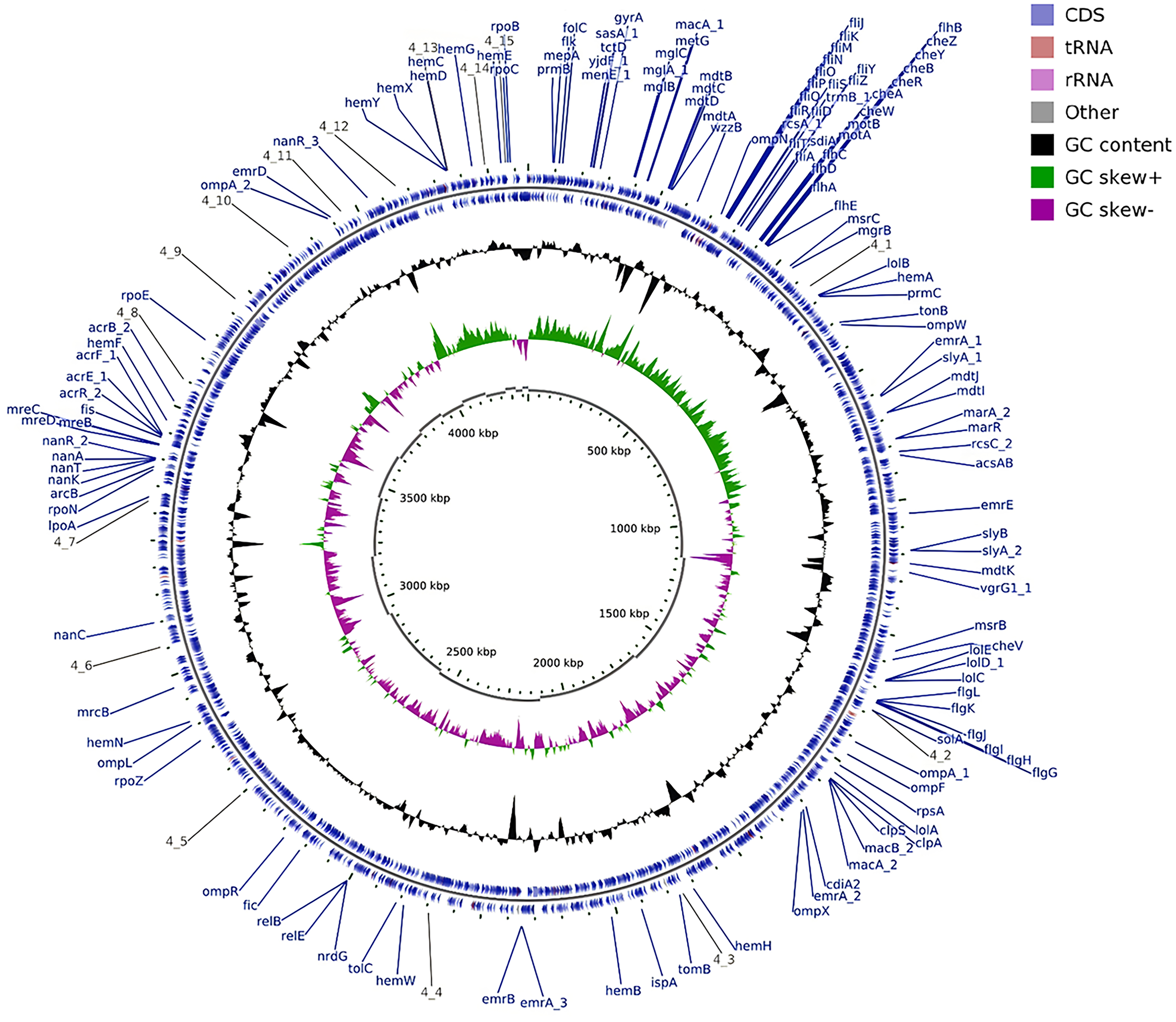
Circular genome of Cronobacter sakazakii strain C4 (novel ST1008). Genome annotation performed using Prokka software. Contigs larger than 20 kb were visualized using CGView. Representative genes are labeled in blue on the outermost circle.
Notably, ST1, ST8, ST17, and ST40 matched clinical isolates from China and global (Lepuschitz et al., 2019), suggesting potential cross-transmission between food and clinical settings. In contrast, ST58, ST226, and ST1008 were absent in Chinese clinical reported isolates, which contrasts with their clinical prevalence worldwide, contrasting with their sporadic global distribution and implying possible niche adaptation to food matrices or environmental reservoirs. Future epidemiological investigations should prioritize trace-back studies and transmission pattern analysis to elucidate potential foodborne transmission pathways.
Virulence gene profiling
Beyond prevalence assessment, 42 virulence-related genes in C. sakazakii isolates were identified by WGS. In this study, 10 C. sakazakii isolates were characterized focusing on the distribution of key virulence gene families. The identified genes were categorized into five functional groups: (1) mobility, invasion, and adhesion genes (rcsAB, flgA, flgB, mviN), (2) outer membrane protein genes (lolA, motB, ompA, ompX), (3) chemotaxis genes (tonB, cheR, cheY), (4) toxin–antitoxin genes (acrR, emrA, emrB, fic, relB), and (5) tolerance-related genes, including those for acid-base tolerance (rpoS), desiccation tolerance (cheB, wzzB), heat tolerance (hsp20), and salivary acid utilization (nanK) (Parra-Flores et al., 2021). Detailed distribution of these virulence genes is presented in Figure 4.
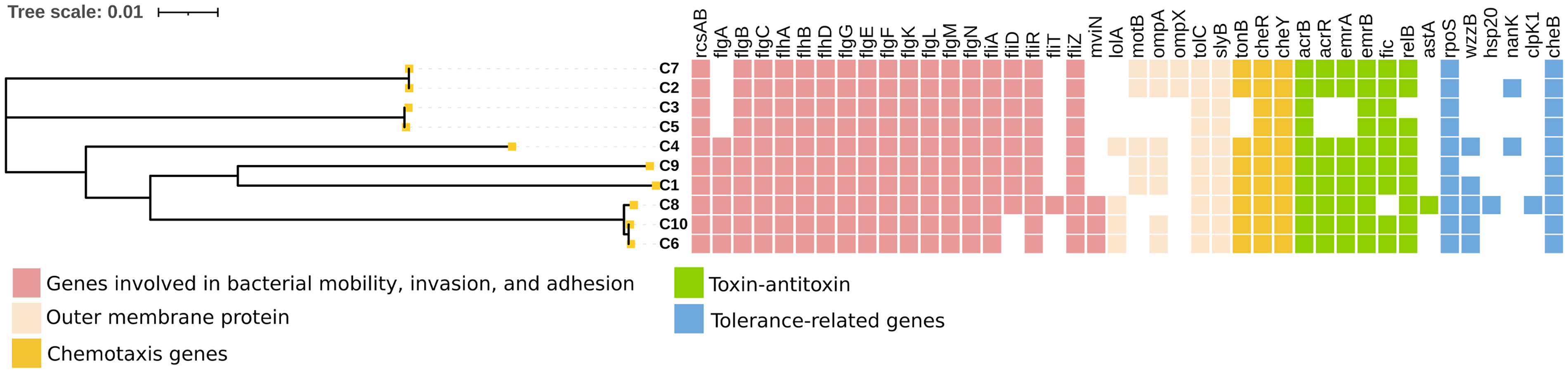
Virulence gene profiles in Cronobacter sakazakii genomes. Virulence genes identified in 10 C. sakazakii strains using Prokka software. Colored squares represent predicted virulent genes, categorized into five functional regions.
The flagellar system, represented by over 20 genes including flgA, fliD, fliT, and mviN, is crucial for motility and host invasion, with flgK specifically regulating motility and stress response. The variable distribution of membrane-associated virulence factors suggests diverse pathogenic potentials. The synergy between ompA and invasion mechanisms, along with stress response mechanisms like the Fic/RelB toxin–antitoxin system, highlights the adaptive capabilities of these isolates. These findings align with previous studies while revealing distinct patterns among isolates from Nanning City, warranting further investigation into their pathogenic mechanisms (Chandrapala et al., 2014; Hill et al., 2021; Hu et al., 2024; Li et al., 2023).
Antibiotic resistance and resistance gene profiling
The study identified 22 distinct resistance-associated genes, including mcr-9 (colistin resistance) and qacJ (disinfectant tolerance) (Fig. 5). All isolates (10/10) exhibited resistance to selected β-lactams (amoxicillin/clavulanic acid, ampicillin/sulbactam, cefoxitin, and Cefazolin) (Table 2). Antibiotics Resistance Genes(ARGs)analysis revealed a complex genetic basis for this resistance, with all strains carrying core resistance genes (acrA, adeF, CRP, emrB) while showing variable distribution of qacG (90.0%), EF-tu (70.0%), and mcr-9 (10.0%) (Jaradat et al., 2022). Notably, the ST1008 isolates uniquely harbored qacJ and CSA-2 genes, distinguishing them from other strains. The qacJ gene showed a strong correlation with the minimum inhibitory concentration of disinfectants (El Sayed Zaki et al., 2019). Additionally, strain C8 exhibited intermediate resistance to chloramphenicol and minocycline, while strain C1 showed intermediate resistance to tigecycline. Moreover, six isolates (C1, C2, C3, C4, C5, and C9) displayed intermediate resistance to cefuroxime. All isolates remained susceptible to the remaining 19 antimicrobial agents tested (Table 2).
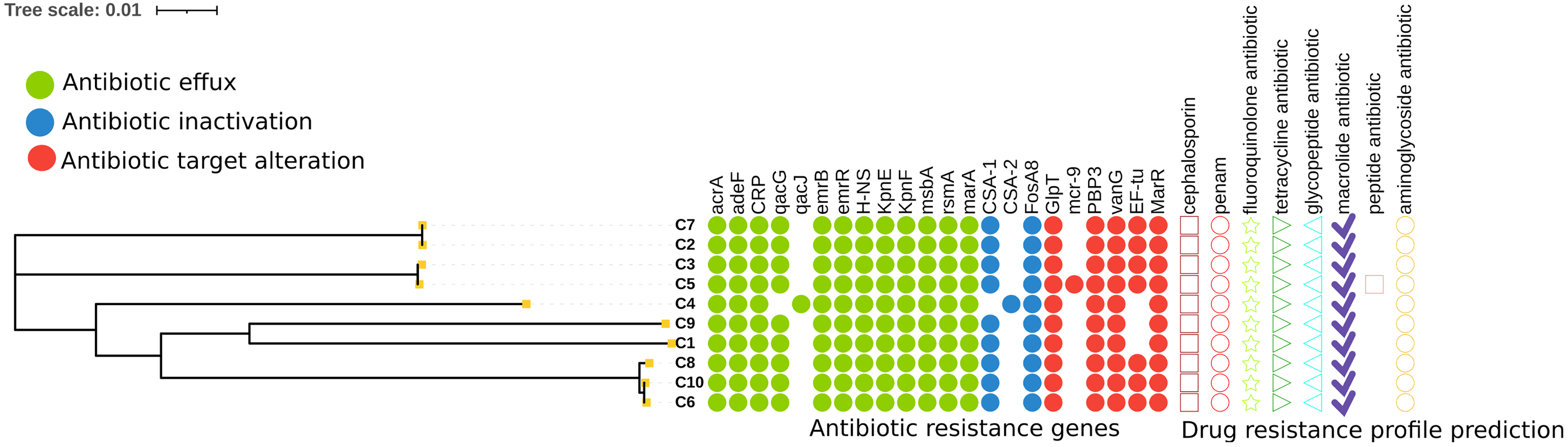
Antibiotic resistance gene profiles in Cronobacter sakazakii genomes. The antibiotic resistance genes were identified by the Comprehensive Antibiotic Resistance Database (CARD). Colored dots indicate predicted resistance genes, classified by resistance mechanisms across three regions. Distinct markers represent specific drug resistance profiles.
Antimicrobial Susceptibility of 10 Cronobacter sakazakii Isolates to 27 Antimicrobial Agents
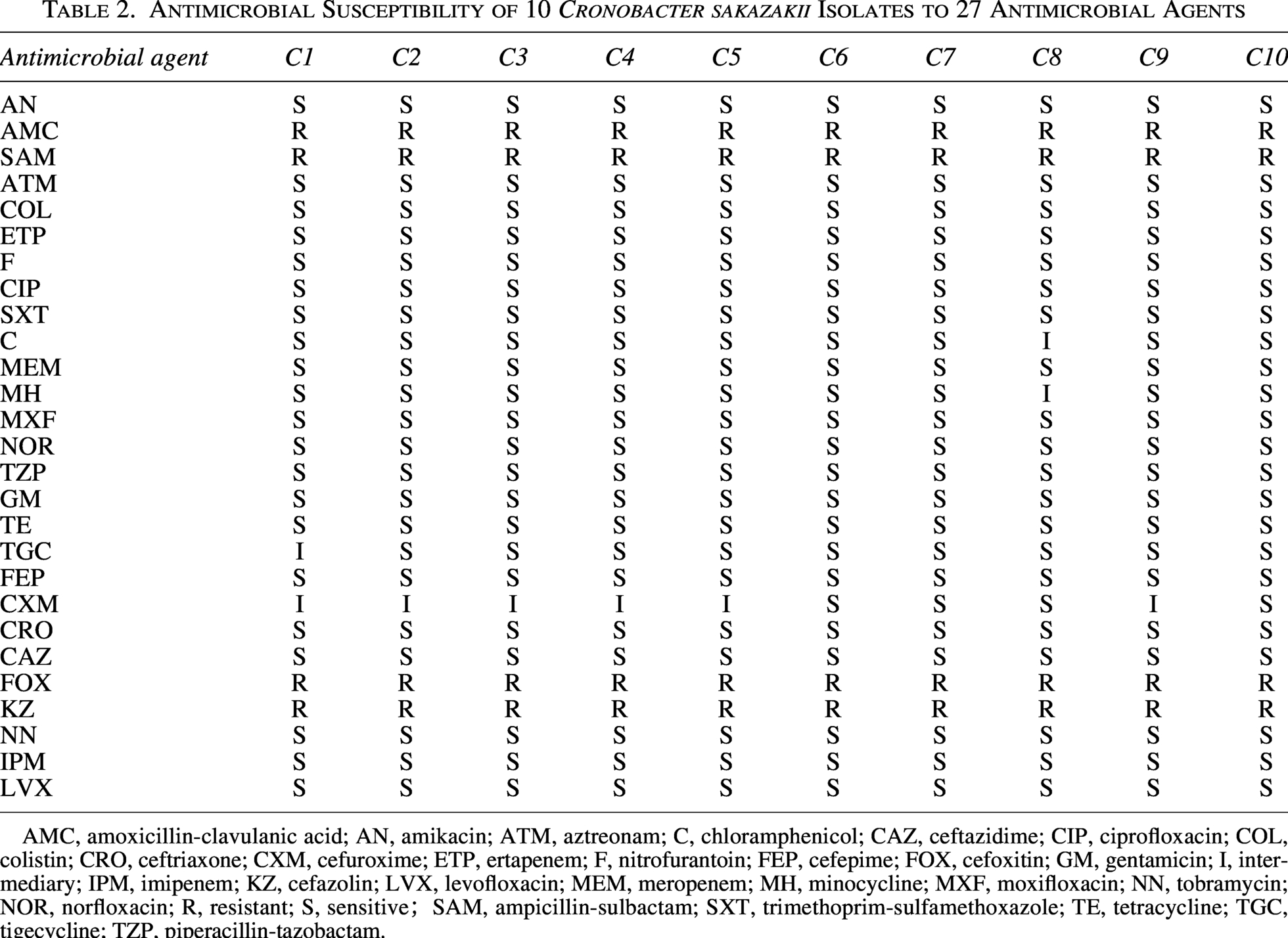
AMC, amoxicillin-clavulanic acid; AN, amikacin; ATM, aztreonam; C, chloramphenicol; CAZ, ceftazidime; CIP, ciprofloxacin; COL, colistin; CRO, ceftriaxone; CXM, cefuroxime; ETP, ertapenem; F, nitrofurantoin; FEP, cefepime; FOX, cefoxitin; GM, gentamicin; I, intermediary; IPM, imipenem; KZ, cefazolin; LVX, levofloxacin; MEM, meropenem; MH, minocycline; MXF, moxifloxacin; NN, tobramycin; NOR, norfloxacin; R, resistant; S, sensitive;SAM, ampicillin-sulbactam; SXT, trimethoprim-sulfamethoxazole; TE, tetracycline; TGC, tigecycline; TZP, piperacillin-tazobactam.
The presence of colistin resistance genes, such as mcr-9, upstream of the IS26 in isolate C5, indicating mobility potential (Song et al., 2024). Despite this genotypic detection, phenotypic susceptibility testing revealed colistin sensitivity across all isolates. Such discrepancies between genotypic predictions and phenotypic susceptibility align with established mechanisms in Cronobacter and related Enterobacteriaceae, where regulatory networks (e.g., H-NS-mediated silencing) can suppress resistance gene expression despite their genetic presence (Cooper et al., 2024). Similar patterns were observed in infant formula isolates (Fei et al., 2022), but the elevated prevalence of these “silent” resistance genes in raw powdered spices with instant cereals suggests unique environmental pressures (e.g., desiccation stress) may amplify regulatory effects in low-moisture matrices (Wang et al., 2024).
Plasmid- and mobile genetic element-mediated dissemination of resistance
Plasmid analysis revealed Col440I (40.0%, 4/10) and IncFIB (pCTU3) (20.0%, 2/10) as key vectors for resistance dissemination (Table 3). The co-occurrence of virulence and resistance genes on mobile elements (e.g., IncFIB plasmids) highlights risks of horizontal gene transfer (Holt et al., 2023). The colistin resistance gene mcr-9, frequently associated with Enterobacter species, was detected upstream of an IS26 element in isolate C5, posing significant public health implications due to its association with multidrug-resistant Gram-negative pathogens (Li et al., 2020; Song et al., 2024). In isolate C8, the heat shock survival family ATPase gene ClpK1, which facilitates bacterial stress response and survival (Guragain et al., 2021), is located within the IncFIB (pCTU3) plasmid. Similarly noteworthy is that the astA gene, which encodes the EAST-1 enterotoxin commonly found in pathogenic E. coli variants, was detected to form downstream of an ISEic2 (Hu et al., 2021; Paiva De Sousa and Dubreuil, 2001). The heterogeneous distribution of these MGEs among isolates, even those sharing the same sequence type, suggests a complex mechanism underlying C. sakazakii virulence dissemination.
Plasmids and Mobile Genetic Elements in Cronobacter sakazakii Genomes

—, Not detected.
The high prevalence of virulent and antibiotic-resistant C. sakazakii in retail spices and cereals poses significant risks, particularly for vulnerable populations (e.g., children, elderly, immunocompromised individuals). Immediate implementation of thermal processing (>70°C) and enhanced hygiene protocols (Arku et al., 2011) are criticato mitigate contamination. However, the current findings are constrained by limited sample size and insufficient tracing of contamination sources within production chains. To address these challenges, future research should adopt multidisciplinary approaches to advance risk assessment models and facilitate the development of targeted interventions for enhanced food safety assurance.
Conclusion
In conclusion, this study reveals significant C. sakazakii contamination in retail powdered spices and cereals in China, with novel strains ST1008 and coexisting resistance-virulence traits identified. The dominance of clinical-associated serotype O1/ST1 highlights foodborne transmission risks. Enhanced surveillance and targeted controls are critical to protect vulnerable populations.
Authors’ Contributions
Writing: L.Z., H.S., K.B., J.Q., and T.W. Investigation: L.Z., H.Y., Z.H., and X.L. Resources: H.S., J.Q., and H.N. Software: L.Z., P.L., and S.W.
Footnotes
Acknowledgment
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This research was funded by the Natural Science Fund Project of Guangxi Province of China (2023GXNSFAA026038) and the Health Commission of Guangxi Zhuang Autonomous Region (Z20200045).
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.