
Abstract
Abstract
Background:
Breathlessness at rest or on minimal exertion despite optimal treatment of underlying cause(s) is distressing and prevalent. Opioids can reduce the intensity of chronic refractory breathlessness and an anxiolytic may be of benefit. This pilot aimed to determine the safety and feasibility of conducting a phase III study on the intensity of breathlessness by adding regular benzodiazepine to low-dose opioid.
Methods:
This is a single site, open label phase II study of the addition of regular clonazepam 0.5 mg nocte orally to KapanolR 10 mg (sustained release morphine sulphate) orally mane together with docusate/sennosides in people with modified Medical Research Council Scale ≥2. Breathlessness intensity on day four was the efficacy outcome. Participants could extend for another 10 days if they achieved >15% reduction over their own baseline breathlessness intensity.
Results:
Eleven people had trial medication (eight males, median age 78 years (68 to 89); all had COPD; median Karnofsky 70 (50 to 80); six were on long-term home oxygen. Ten people completed day four. One person withdrew because of unsteadiness on day four. Five participants reached the 15% reduction, but only three went on to the extension study, all completing without toxicity.
Conclusion:
This study was safe, feasible and there appears to be a group who derive benefits comparable to titrated opioids. Given the widespread use of benzodiazepines for the symptomatic treatment of chronic refractory breathlessness and its poor evidence base, there is justification for a definitive phase III study.
Introduction
Regular, low-dose opioids can safely reduce the intensity of chronic refractory breathlessness.5–7 Opioids are useful agents to palliate dyspnea, but there is a need to have nonopioid medications to augment the benefits of opioids or as a substitute for people who cannot tolerate them or derive no benefit from them.
Although anxiety is rarely the sole cause of breathlessness, 8 it is implicated in the worsening and maintenance of dyspnea. A medication with an anxiolytic effect is a logical choice to explore in prospective clinical studies. Despite widespread use of benzodiazepines for palliation of chronic refractory breathlessness in hospice/palliative care, a recent Cochrane review concluded that there were insufficient data to recommend their use in this setting, 9 and even fewer data are available for the combination of opioids and benzodiazepines. 10 The only sizable study was in people in the last hours or days of life and cannot be applied to people earlier in their disease trajectories. 11
A long-acting benzodiazepine with pharmacodynamic and pharmacokinetic data for a range of formulations is the ideal choice. Clonazepam is the benzodiazepine with the widest choice of formulations (tablet, liquid, injectable) and once daily dosing.
Given the evidence of the efficacy of morphine for the relief of breathlessness, the aim of this pilot study was to determine whether it is safe and feasible to conduct a fully powered phase III study of the addition of clonazepam to regular low-dose sustained release morphine for the reduction of chronic refractory breathlessness. The primary outcome in this pilot study was safety (drowsiness, respiratory depression, worsening cognition). Secondary outcomes were feasibility (recruitment and completion rates, and the logistics for running the phase III study) and efficacy (any evidence of net benefit over baseline).
Methods
Study design
This is a single site, open label phase II study of commencing regular clonazepam 0.5 mg nocte orally with KapanolR 10 mg (sustained release morphine) mane orally and docusate and sennosides in people with chronic refractory breathlessness who are naive to opioids and benzodiazepines. Refractory breathlessness was where all reversible causes for breathlessness had been optimally treated. 6 The primary outcome was assessed after four days of therapy. Participants could choose to extend using the two medications for another 10 days if they achieved >15% reduction in dyspnea intensity over their own baseline.
Study participants
The study intended to have complete data for four days on 10 opioid-naïve outpatients who had a palliative diagnosis, aged 18 years or more who scored ≥2 on the modified Medical Research Council (mMRC) scale,12,13 on stable medications, and a prognosis of >2 months.
Exclusion criteria included adverse reactions to study medications; already using study medications at or above study doses; on a monoamine oxidase inhibitor; poor cognition (Folstein Mini-mental Status Exam <24/30); 14 Australian-modified Karnofsky Perfomance Status (AKPS) <50 at baseline; 15 uncontrolled nausea, vomiting, and/or gastrointestinal obstruction; estimated glomerular filtration rate <25 mls/minute; 16 serum alkaline phosphatase, bilirubin, ALT, or AST >3x the upper limit of normal; resting respiratory rate <8; respiratory or cardiac event in the previous week (excluding upper respiratory tract infections); pregnancy; or unable to give informed consent or complete diary entries. Participants did not need to have anxiety to qualify for the study.
Settings
Participants were recruited between October 2011 and February 2012 from a regional palliative care service that provides inpatient, consultative, outpatient, and community care in the public and private sector.
Measurements
Safety
Safety assessments included 100 mm visual analogue scales (VAS) for drowsiness, worsening cognition, or respiratory depression; and monitoring oxygen saturation (SaO2 ) and end-tidal CO2 at baseline, the end of day 5, and for those who entered the extension, at the end of day 14. These measurements used the LifeSense LS1-9R (Nonin Medical Inc., Delsbo, Sweden).
Feasibility
Feasibility measures were: reasons for nonrecruitment at screening; recruitment and completion rates; acceptability of patient diaries; and qualitative feedback from study staff about the logistics of running a fully powered study.
Efficacy
The primary outcome measure to inform the power calculations of the phase III study was based on breathlessness right now (morning and evening) on the VAS on day four. Secondary efficacy outcomes include worst, best, and average breathlessness in the previous 12 hours (measured morning and evening).
Data collection and data quality
Demographic and clinical data were collected by research staff. Patients filled out twice daily diaries with VAS scores for breathlessness (now, worst average, and best in the last 12 hours); constipation; and sleep quality.
Sample size
Data on 10 participants was required to calculate the effect size to inform a power calculation for the phase III study.
Analysis
Safety, feasibility, and efficacy were summarized descriptively.
Ethics, consent, and trial registration
The study was approved by the Southern Adelaide Clinical Human Research and Ethics Committees and registered with the Australian Clinical Trial Register (ACTRN12610000589088). All participants provided written informed consent.
Results
Participant flow and study population
Twenty people with dyspnea were screened. The major reasons for not progressing from screening to consent were: already on opioids (n=3), AKPS<50 (n=2), out of geographic region (n=1), and death (n=1) (Fig. 1).
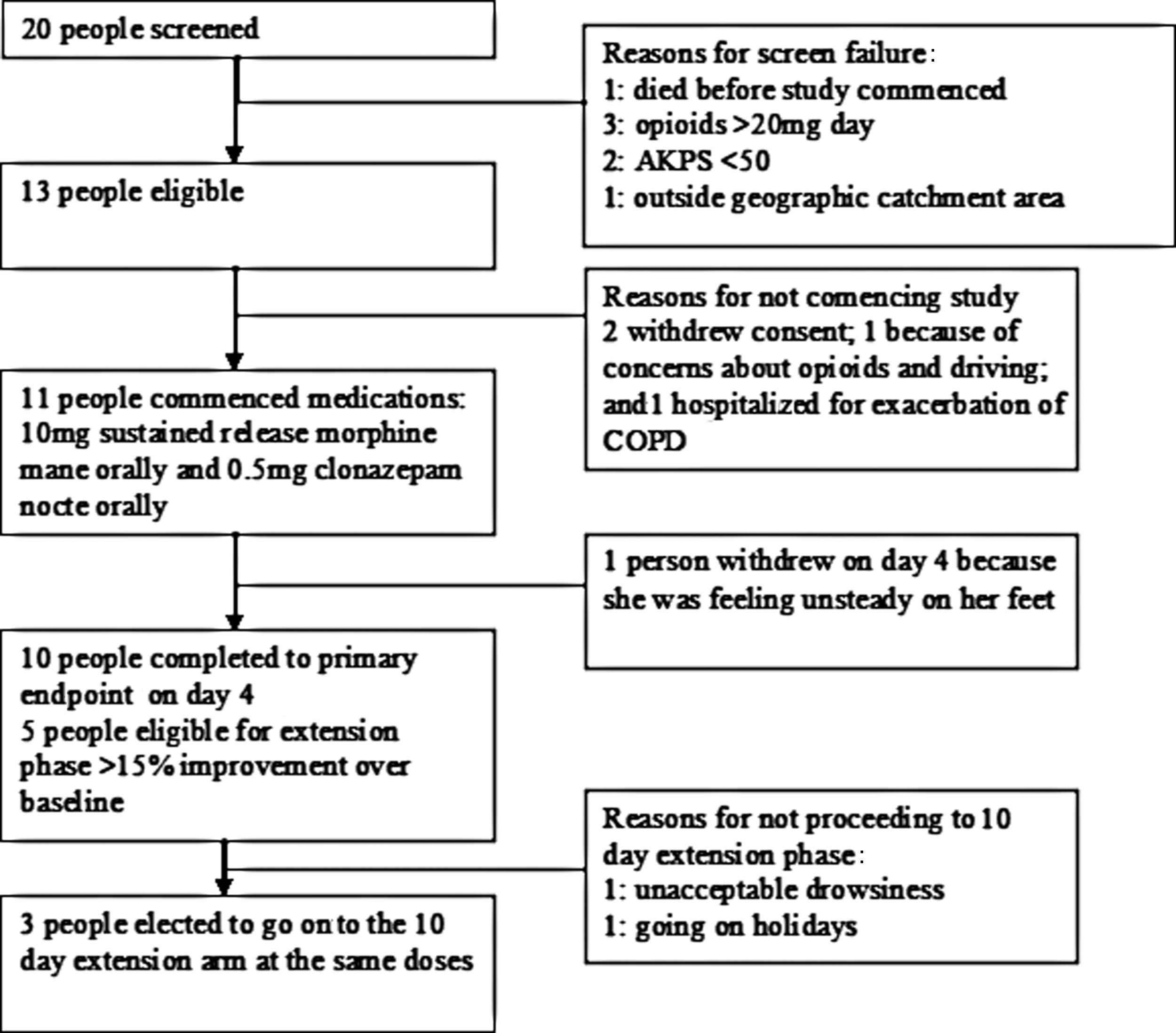
Participant flow. (COPD, chronic obstructive pulmonary disease; AKPS, Australian-modified Karnofsky performance status.)
Thirteen people consented for the study and 11 had medication dispensed (8 males, median age 78 years (68 to 89); all had COPD and AKPS median 70 (50 to 80); 6 were on long-term home oxygen). Vital signs included a median respiratory rate of 19.5 (15 to 27), pulse oximetry 92 (90 to 98), and end-tidal CO2 25 (15 to 38) on room air. One person withdrew before commencing and one was hospitalized before beginning the study. One patient withdrew during the study (day four, continued to feel unsteady on his feet), and 10 completed the study to the end of day four.
At baseline, morning VAS ‘breathlessness right now’ had a median of 68.5 mm (range 31–86). Evening VAS ‘breathlessness right now’ had a median of 63.5 (range 9–75).
For the extension study, five participants achieved a 15% improvement in ‘breathlessness right now’ over baseline levels, of whom three went on to the 10-day extension phase, successfully completing it.
Safety
No participants were hospitalized during the course of the study. No participants were treated for respiratory depression, and end-tidal CO2 remained stable in all participants. By day four, two participants reported feeling ‘much more sleepy or drowsy than usual’ and three more that their sleepiness or drowsiness was ‘more than usual.’ No one reported clouded cognition or confusion. Only one person withdrew after commencing, on day four, because of feeling unsteady walking. Three other people reported this but did not withdraw. One person reported new difficulties with urinary stream. Constipation was not symptomatic for participants.
Feasibility
On average, the study recruited one participant per week from one site from a screening rate of 1.7 participants per week, and was completed in a 12-week period. Ten out of 11 people who started medication completed to day four.
Efficacy
The median score for morning average dyspnea right now was 49.5 (6 to 87) with a median reduction of 9 mm (23 mm worsening to 80 mm improvement) over baseline and in the evening a median of 45.4 (2 to 84) with a median improvement of 6.5 mm (18 mm worsening to 64 mm improvement) over baseline. Quality of sleep showed no change over baseline.
Discussion
These data confirm that it is safe and feasible to proceed to a phase III study using benzodiazepines as an adjunct to opioids in the symptomatic management of breathlessness given their current widespread use. The question of net effect (accounting for both symptomatic benefit and any toxicities) will only be answered by an adequately powered study. The median improvement in morning and evening ‘breathlessness right now’ scores are the same order of magnitude seen in the phase III placebo controlled trials of 20 mg daily of sustained release morphine. 6
Drowsiness was a symptom for five participants, but this may or may not be transient. This will need to be evaluated in future work with longer periods of follow-up.
Five people achieved a 15% reduction over their own baseline dyspnea scores, but only three of these people elected to continue to day 14. Although there are no efficacy conclusions given small numbers, this rate is disappointing.
Recruitment rates for this study were excellent; the work of identifying potential participants and successfully supporting them to completion was done by a team that is skilled in palliative care research, especially in people with chronic refractory breathlessness. A more conservative estimate of recruitment will underpin feasibility calculations as this proceeds to a multisite, phase III study.
What are the other designs that could have been used? The design could have simply been of clonazepam in opioid and benzodiazepine naive participants, but ethically this may have been difficult to defend. The author team has good quality data about the efficacy of opioids alone in controlled clinical trials and in prospective dose ranging studies that could be used to give a historical baseline. There is level-one evidence demonstrating the ability of opioids to reduce chronic refractory breathlessness. The adequately powered randomized controlled trial (RCT) was at 20 mg sustained release morphine per 24 hours. More recent work suggests benefit for many people at 10 mg sustained release morphine per 24 hours. At the same time, a recent systematic review suggests that benzodiazepines did not have evidence of their net benefit. As such, setting the dose of morphine at 10 mg (half that of the RCT) with addition of clonazepam was the compromise made by the authors in designing the study. This was a design decision, and the investigators felt it would have been ethically more difficult to justify only using clonazepam given the recent Cochrane review that suggested there was not enough evidence to support its use alone.
What does a definitive study look like?
There are several design options. Given evidence that opioids are effective in chronic refractory dyspnea, option one would be to titrate opioids to a level of benefit, and then, in a double blind, randomized study add either low-dose benzodiazepine or an additional small increment of opioid (to see if additional benefit can be gained). Another option would be to add clonazepam only to people who did not gain net benefit from opioids. A third option would be to compare daily benzodiazepines with opioids in a phase III study. The population would be earlier in the disease than the participants at the end of life in Navigante's study. 11 The primary outcome will also need to be considered carefully. The current pilot used the intensity of breathlessness, but the unpleasantness may arguably also be a reasonable primary outcome.
All participants would complete the Hospital Anxiety and Depression Scale and anxiety scores will be dichotomized in stratification at randomization. Any such study would also need to have an ability to ensure that any early responders maintained benefit over time and that early toxicity related to drowsiness disappeared.
Clinical benefit was experienced in the current study but perhaps at the cost of more toxicity. Carefully collected data on benefit and harms will be needed prospectively for a longer period of follow-up.
Footnotes
Acknowledgments
Thanks go to Ms. Debbie Marriott for her expertise in manuscript formatting and submission, Zac Vandersman for his work with the data and to all of the participants who gave their time. The funding for this study was made available from Basil Thomas Bequest to Repatriation General Hospital, Daw Park, Australia. The generosity of Foundation Daw Park is appreciated.
Author Disclosure Statement
No competing financial interests exist.