
Abstract
Background:
People with sickle cell disease (SCD) have a life expectancy of <50 years, so understanding their end-of-life care is critical.
Objective:
We aimed to determine where individuals with SCD were dying and their patterns of care in the year preceding death to highlight end-of-life research priorities and possible opportunities for intervention.
Design:
Using the California SCD Data Collection Program database (containing administrative data, vital records, and Medicaid claims), we examined people with SCD who died between 2006 and 2015 (cases) at age <80 years and examined their hospital and emergency department (ED) utilization in their last year of life. Comparators included living controls with SCD matched 1:1 based on age, analysis year, insurance, and income.
Results:
We identified 486 people with SCD (cases) who died at a median age of 45 years (SD: 16 years). Most died in the hospital (63%) and ED (15%). In their last year of life, people with SCD were hospitalized for an average of 42 days (SD: 49 days) over five admissions. Inpatient admissions and ED visits were stable throughout the year until the month before death when acute care utilization sharply increased. In their last year of life, cases had more hospitalizations than controls, but similar ED utilization.
Conclusions:
People with SCD are dying acutely at a young age and most die in the hospital and the ED. Since clinicians caring for people with SCD currently cannot predict which acute events may be life-threatening, a comprehensive palliative approach to people with SCD must extend beyond chronic pain management and psychosocial support to include advance care planning.
Introduction
Medical advances such as penicillin prophylaxis, hydroxyurea, and chronic red blood cell exchange allow children with sickle cell disease (SCD) to live well into adulthood; however, adults with SCD continue to demonstrate shortened life expectancy that is well below 50 years.1–6 SCD is an understudied disease as evidenced by legislation currently under consideration to increase funding for SCD research. 7 Despite the call for a palliative approach to people with serious illness,8–12 little is known about end-of-life care for people with SCD. A deeper understanding of issues related to end-of-life care remains of utmost importance for this vulnerable patient population.
Current end-of-life studies in SCD mostly focus on mortality rates, life expectancy predictions, and causes of death.3,13,14 To ensure that individuals with SCD are receiving appropriate goal-directed end-of-life care, we need to know where they are dying and their patterns of health care utilization at end of life. Only then can we determine where and when palliative care, end-of-life, and goals of care interventions are most needed and critically evaluate the effectiveness of a palliative approach to care for people with SCD beyond symptom management. We can also better inform care providers of patterns of illness or health care utilization associated with increased risk of death. We used a retrospective population-based analysis to determine where people with SCD are dying and their patterns of care in the year preceding death.
Methods
Study design and oversight
The California Sickle Cell Data Collection (SCDC) Program aims to identify people with SCD in California through a multisource database comprising data from (1) the Office of Statewide Health Planning and Development administrative data sources (hospital patient discharge dataset [PDD] and emergency department [ED]), (2) vital records, and (3) Medicaid claims. The SCDC then links all data by person and applies a validated case definition for SCD to identify a statewide cohort spanning 2004 through 2015. 15
Individuals with SCD are identified by the presence of three or more separate health care encounters over five years with SCD ICD-9/10-CM. 16 Additionally, because of the small number, a lower likelihood of reaching advanced age, and a higher rate of health care utilization not associated with SCD than the remainder of the cohort, those over the age of 70 are reviewed individually and in depth to determine whether there is a possibility of coding errors such as repeat coding for SCD by a laboratory when sickle cell trait is the correct diagnosis. The databases and linkage processes have been previously described.17,18 The SCDC and this current study were overseen by the California Committee for the Protection of Human Subjects.
Patient population
Cases
Deceased cohort cases comprised people with SCD who died between 2006 and 2015. Individuals were excluded if their cause of death was due to trauma or peripartum events (either as coded during a terminal admission or on the death certificate) or if no record linkage between the vital records could be made. We adopted this approach to increase our likelihood for analyzing SCD-related causes of death rather than a sudden terminal event such as trauma (Fig. 1).
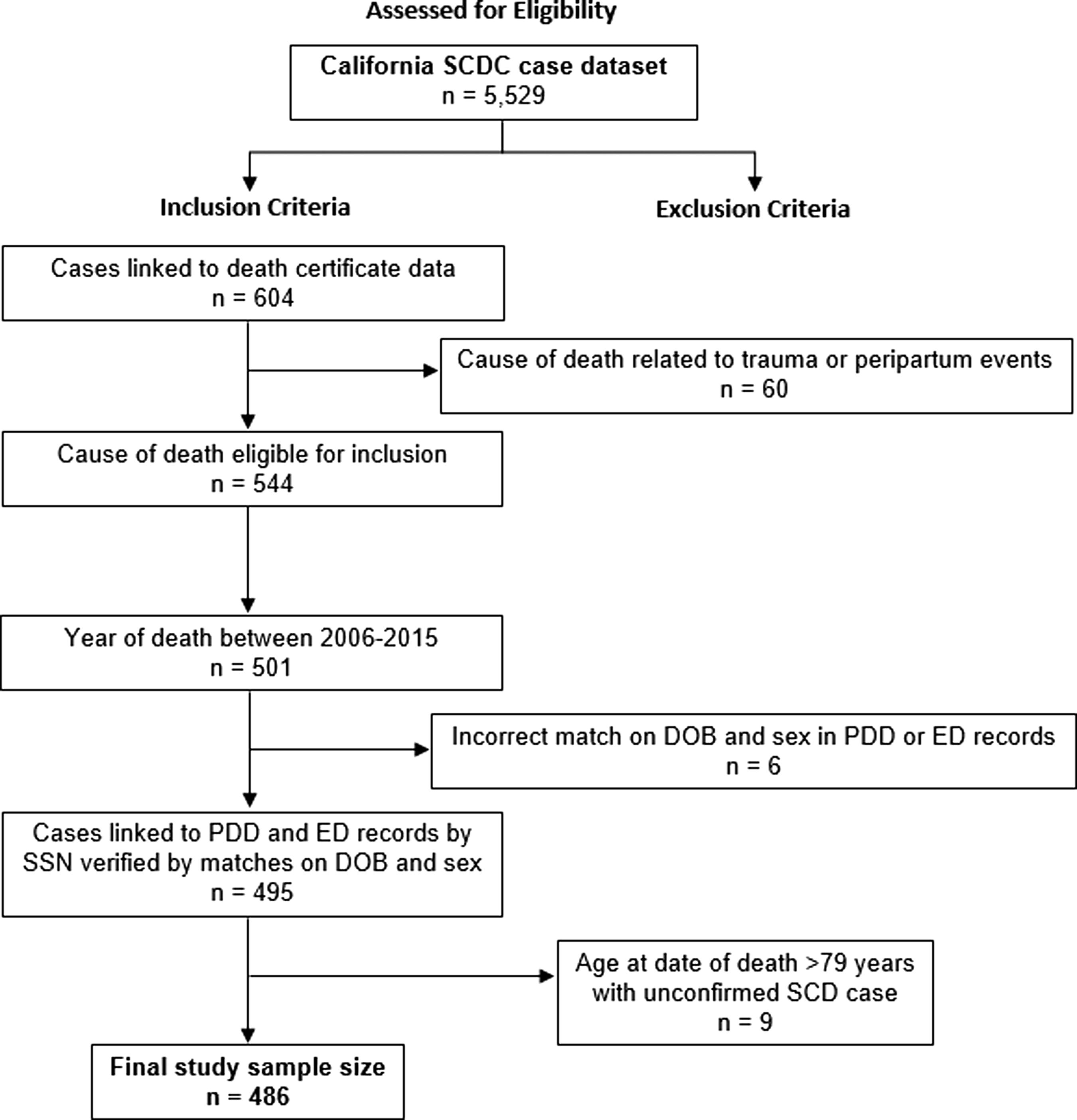
Consort diagram of study population.
Controls
Living controls included people from the SCDC database, who were randomly selected and matched using the 1:1 process to deceased cases by age on date of death of the index case, sex, insurance type, and median income tertile. By anchoring matches to the deceased cases' date of death, we increased our ability to compare health care utilization between matched living and deceased patients during the same 12-month period and decreased the potential for temporal differences in health care utilization, income, and changes in standard of care, etc.
Patient-level measures
We obtained age, race/ethnicity, location of death, and cause of death from the vital statistics death record. We identified insurance type from the payer category listed in the last patient discharge database record before death. When no patient discharge database records existed for a patient, we used the payer category listed in the last ED record.
We estimated socioeconomic status using the median household income of a patient's last residential zip code recorded in the administrative data. Median household income data were obtained from the five-year American Community Surveys (ACSs) from 2011 to 2016 as follows: 2011 ACS data for patients who died between 2005 and 2011 and 2016 ACS data for those who died between 2012 and 2015. We converted ACS data from census tract level to zip code level using the Department of Housing and Urban Development crosswalk data files and guidelines. 19 We then assigned the study population to quantile ranks based on the last median household income.
Health care utilization
The number of hospitalizations, days admitted (PDD records), and treat-and-release ED visits (ED records) in the preceding 12 months were calculated for both cases and controls. Sickle cell specialty care centers meet the California Department of Health Care Services' criteria for care, as defined in the California Children's Services program (most include pediatric and adult services), 20 and patients were categorized as either receiving care at a specialty center in the 12 months before death or not. Intensive care unit (ICU) admissions were identified with codes for intubation, mechanical ventilation, invasive hemodynamic monitoring through arterial catheterization, or central venous catheter monitoring. 21
Statistical analyses
Descriptive statistics were calculated for each patient characteristic and health care utilization measure above. Categorical variables were compared using chi-squared analysis and continuous variables using a t test or ANOVA. The analyses were done using SAS, version 9.4.
Results
Study population
For the 486 people with SCD who died, the mean age at death was 44.1 years (SD: 15.6). Approximately 60% of them died at age 41 years or older (Table 1). Most people with SCD (83%) were publicly insured and 45% lived in neighborhoods with a median household income of less than $50,314. Matched SCD controls (n = 486) had similar demographic characteristics.
Demographics of Cases with Sickle Cell Disease Who Died during the Period 2006–2015 and Matched Controls
Location of death
Most SCD cases died in hospitals (63%) (Fig. 2). A comparable number of SCD cases died in the ED (15%) and at home (14%) and only 0.4% died in hospice. Young adults (22–40 years) had the highest rates of ED deaths (20%), followed by children (0–21 years: 17%) and older adults (≥41 years: 12%). The majority of patients who died in the hospital or emergency room died after short admissions: 57% were admitted for ≤3 days, which did not vary by age.
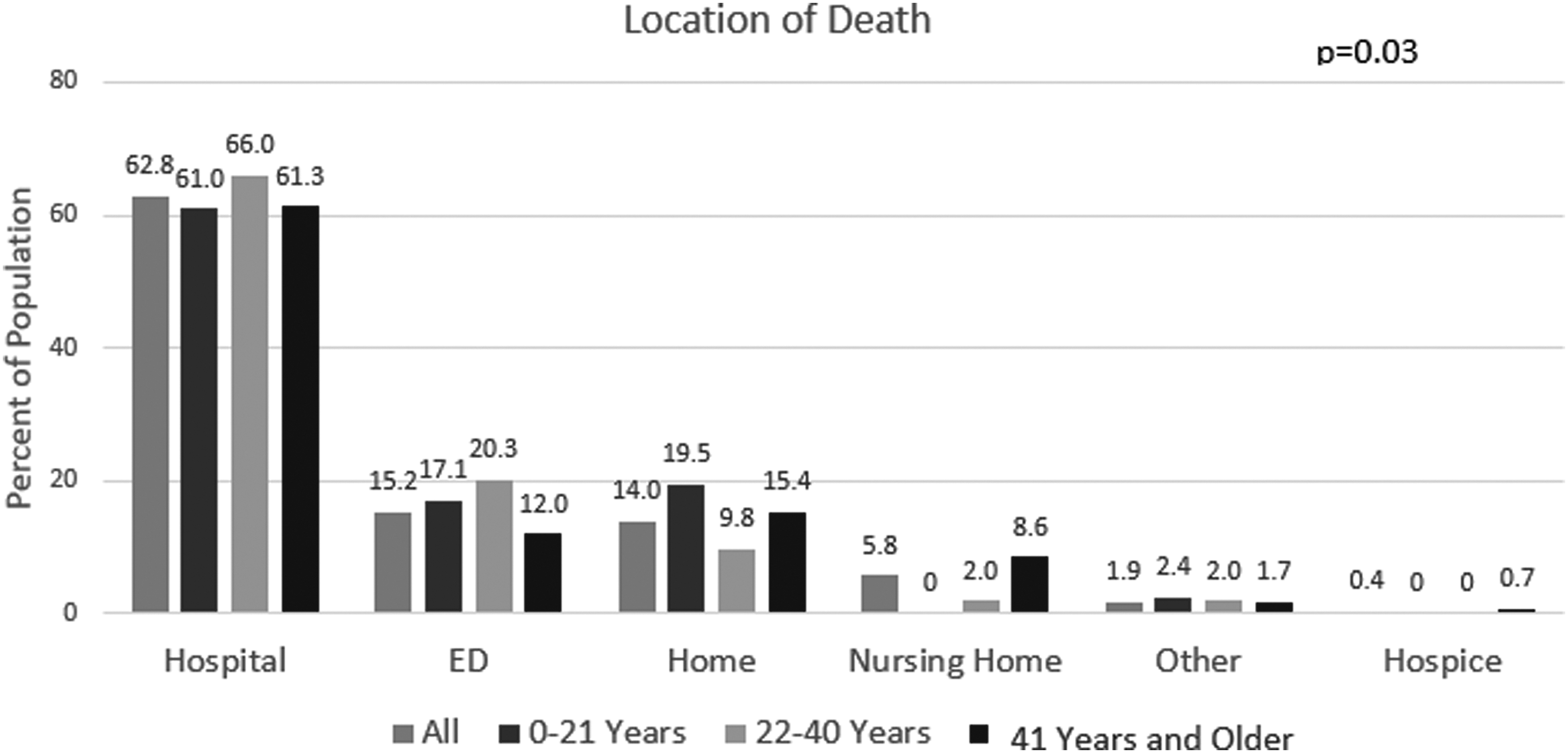
Location of death of people with sickle cell disease by age.
Care in the last year before death
Hospital admissions
The number of hospital admissions, number of ED visits, and ICU utilization rates were stable throughout the last year of life until the month before death when rates of all three parameters rose sharply (Fig. 3). The number of days in the hospital steadily increased throughout the last year before death and did not vary when case utilization was further stratified by insurance and age at death (Fig. 4). Deceased cases had more frequent hospital admissions (5.3 admissions) and days admitted (41.8 days) than living controls (1.6 admissions, p < 0.0001: 8.9 days, p < 0.0001) (Fig. 5). While the number of admissions in the last year of life did not vary by age (p = 0.22), the number of days admitted did: deceased children spent more days admitted in the last year (59.0 days) compared with young adults (43.5 days) or older adults (38.6 days, p = 0.04).
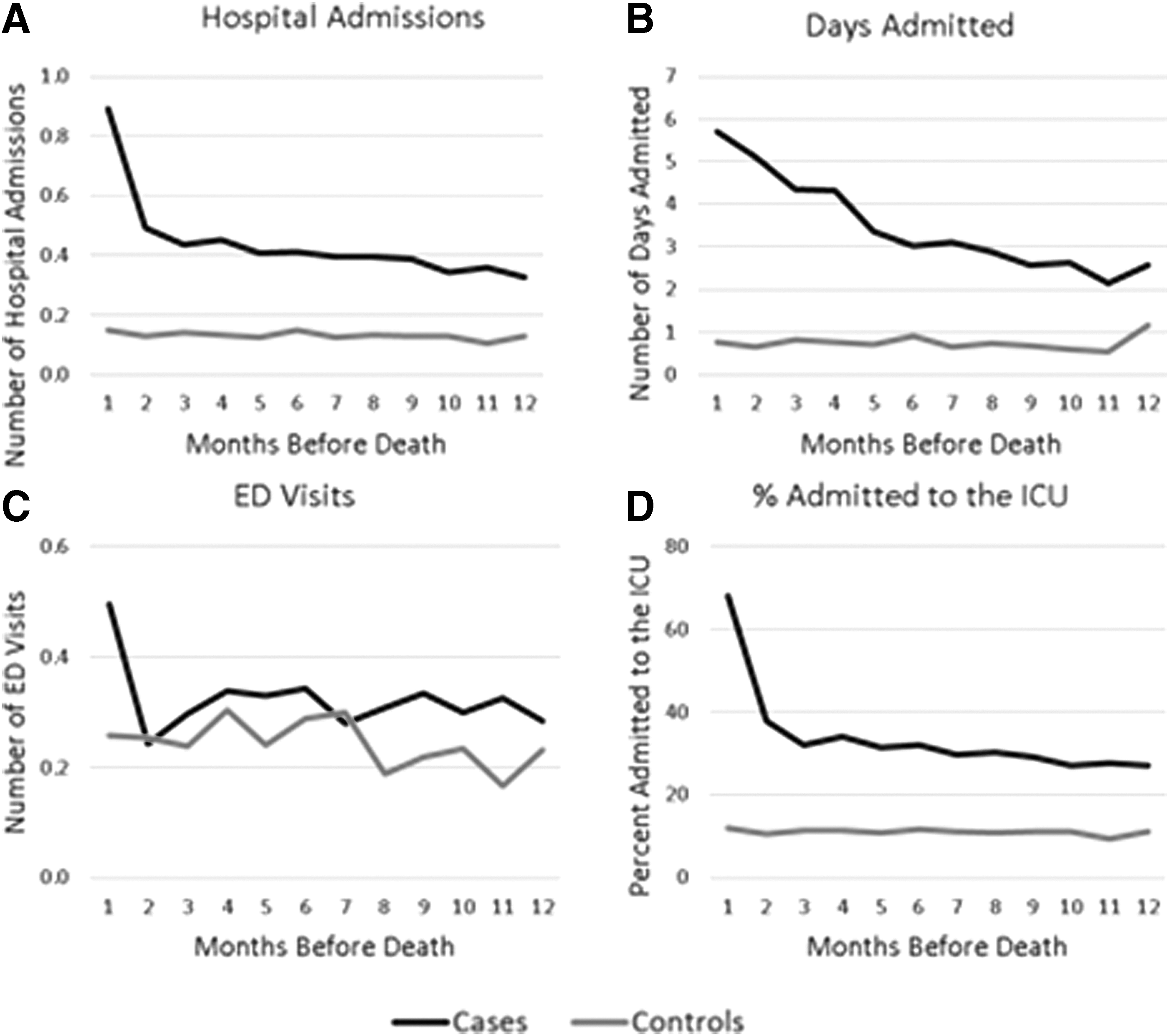
Patterns of care in the preceding year by month: cases versus controls.

Patterns of care in the year before death by month: by insurance and age.
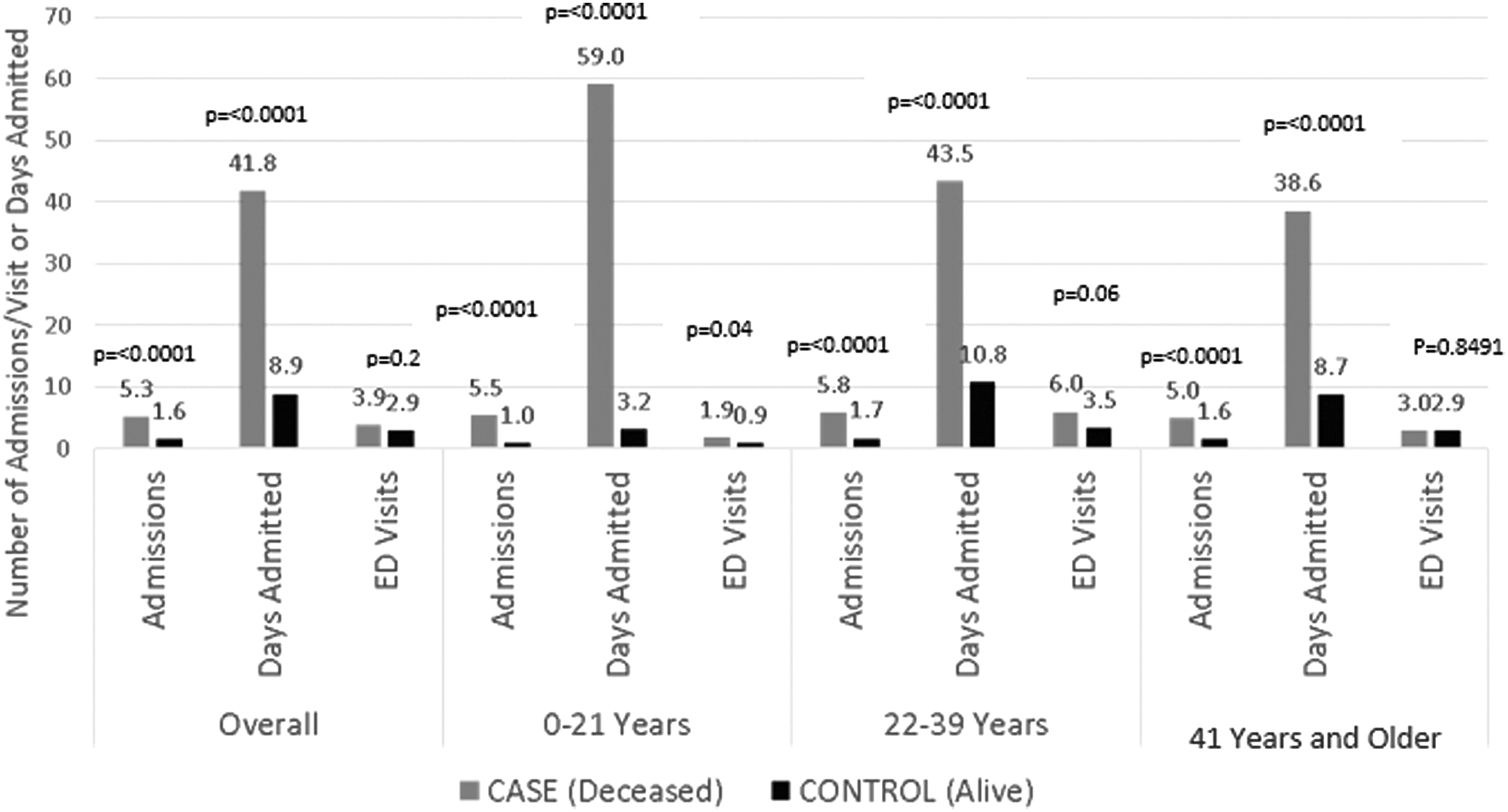
Acute care utilization in the prior year: cases versus controls by age.
ED visits
The number of ED visits was stable throughout the last year of life until the last month of life, when ED utilization frequency rose (Fig. 3). Despite this increase in case ED utilization at end of life, differences in total number of ED visits between deceased cases and living controls did not reach statistical significance (cases: 3.9 visits, controls: 2.9 visits, p = 0.2) in the last year (Fig. 5). Young adult cases had more ED visits (6.0 visits) compared with children (1.9 visits) and adults (3.0 visits, p = 0.01); ED visits did not vary by age in the controls (children: 0.9 visits, young adults: 3.5 visits, and adults: 2.9 visits, p = 0.08).
ICU utilization
The number of ICU visits was stable throughout the last year of life until the last month when rates rose sharply (Fig. 3).
Location of care
Children with SCD were more likely to be seen at a specialty center than the older age groups. Less than 10% of adult admissions occurred at specialty centers (Fig. 6).

Specialty center admission patterns in the year before death by month: cases versus controls and by age.
Discussion
Our study finds that people with SCD are dying acutely; they die in the hospital after short admissions or in the ED and very few die in hospice. In the year before death, deceased individuals have more hospital admissions and are admitted for more days than controls. However, the number of treat-and-release ED visits did not differ between the deceased and living controls. Our findings are consistent with previous studies of causes of death in people with SCD, which show that most die of acute complications and can be relatively healthy before that fatal episode.3,13,14 Therefore, people with SCD and their caregivers live with the fear of sudden death at a young age, particularly in those who are frequently admitted to the hospital. Additionally, most adults with SCD receive care in non-SCD specialty centers. These findings have implications for models of care, particularly surrounding a palliative approach to end-of-life care, and research priorities in SCD.
As above, we found that most individuals with SCD are dying in the hospital after brief admissions or in the ED. Our results are comparable with a previous study of national death certificate data, which found 69% of SCD deaths occurring in the hospital and 16% in the ED. 1 The acuity of SCD deaths highlights the critical importance of incorporating a palliative approach to comprehensive SCD care, including around guidance with end-of-life planning. Palliative care in SCD has mainly focused on management of chronic pain and psychosocial challenges,22–26 particularly now that people with chronic SCD pain are negatively impacted by the opioid epidemic.27–29 As crucial as these areas of need are, our study highlights another equally important reason palliative care principles are needed in SCD care.
Young people with SCD face the distinct reality of dying unexpectedly, so it is imperative that they continue to receive comprehensive care from their hematologists and primary care providers, in addition to consultative care with palliative specialists for advance care planning, 26 wills with guardianship clauses, and opportunities to discuss their legacy. We need to determine whether SCD programs are incorporating best practices to address ongoing pain management and end-of-life issues in young people with SCD.
Given the limited specialized palliative care resources and lack of risk prediction models for early death in people with SCD, initial research should focus on (1) determining what proportion of patients with SCD are completing advance care plans and (2) determining how, when, and which patients with SCD would most benefit from integrating advance care planning with comprehensive SCD care, for example, high hospitalization rates or frequent emergency room visits and those with uncontrolled pain. While we continue to work on risk predication models for early death, we should not neglect the need to improve end-of-life care for these vulnerable patients.
Long-term palliative care exists for many children with complex chronic conditions with expected, but unpredictable, early death. These critically ill children continue to get their primary diseases managed by subspecialists, but have palliative care for symptom management and goals of care support. 30 Perhaps models of integrating specialty and primary palliative care in children with complex conditions could inform how best to integrate palliative care into comprehensive SCD care.
Less than 10% of admissions in the last year of life occurred at specialty centers. Thus, the timing of advance care planning discussions and the location of care for young adults with SCD are important factors that should be carefully weighed when determining best practices of integrating specialized palliative care and advance care planning into SCD care. Community centers caring for individuals with SCD need to be well versed in both management of acute complications of SCD and advance care planning, the latter in collaboration with palliative care specialists.
In oncology, adolescents and young adults (AYAs) admitted at community centers receive different end-of-life care than AYAs admitted at specialty centers. 21 Location of care also affects care for people with SCD. Smaller centers are associated with higher mortality rates for people with SCD, 31 and AYAs with SCD admitted at community centers are more likely to be intubated and have longer lengths of stay than those admitted to pediatric centers. 32 Even among children's hospitals, high volumes of SCD admissions were associated with fewer ICU admissions. 33 Since location of care also impacts end-of-life care for people with SCD, ongoing research and policy measures should prioritize optimizing the quality of care that people with SCD receive during acute illnesses and at end of life at both specialty centers and community hospitals.
Young adults with SCD who died had more ED visits than children and adults with SCD who died, but ED utilization rates did not vary by age in the controls. Many studies have shown that young adults with SCD have high acute care utilization, in lieu of routine hematologic care,34–40 which results in fragmented care 41 and increased risk of mortality.1,6,39 As SCD centers develop transitional and AYA programs, we need to determine how to best integrate palliative care (primary or specialty) into comprehensive SCD care. The pediatric-to-adult transition is challenging for patients with chronic diseases, 42 with our study raising even more concerns that young adults with SCD frequenting the ED are dying at higher rates. Is this subgroup of young adults dying of SCD driving the increased acute care utilization seen in young adults with SCD? Are they higher utilizers because they have always been sicker or because they lack sufficient preventative and supportive care? Would more comprehensive SCD care during the transition period prevent their increased acute care utilization and early death? Developing risk prediction models for early mortality for patients with SCD should be a research priority and can help guide the timing of advance care planning and specialty palliative care integration into the SCD care model.
Since most people with SCD are publicly insured, changes in best practices and care models for SCD have policy implications.38,40 Medicaid and other insurers must make end-of-life needs in SCD a priority. In particular, sufficient reimbursement for palliative care and advance care planning is essential. Cuts to Medicare/Medicaid reimbursement rates for complicated visits could severely hurt SCD patients, especially as this study shows they too need to engage in critical advance care planning conversations.
Since SCD is an orphan disease, government support and incentives for SCD research, including the proposed end-of-life studies, are crucial. This article highlights five important areas for future SCD research: (1) developing risk prediction models for early mortality for patients with SCD; (2) determining if patients with SCD are completing advance care plans; (3) how and when to integrate advance care planning and specialty palliative care into SCD care—potentially incorporating the aforementioned risk prediction models; (4) how to better serve high utilizing AYAs; and (5) determining when the increased utilization between cases and controls begins and if that is a moment for possible interventions to decrease SCD mortality.
As with any population-based study, ours has some limitations to consider. Our study population is limited to California, which has fewer people with SCD and strong government safety net programs, compared with many other states. However, we established a methodology that can be used to determine if the same end-of-life care patterns exist outside of California. In addition, hospitalization rates for controls are comparable with multistate studies of acute care utilization.34,37,43–45 We lacked granular data on causes of death, making it difficult to determine if the primary cause of death was an SCD complication as the vast majority of death certificates and terminal admission codes were SCD with or without crisis, which is not a primary cause of death. However, removing trauma and peripartum events does eliminate deaths from nondisease-related causes. Administrative data have been shown to be useful in conducting surveillance in hemoglobinopathies,15,46,47 but significant limitations exist in SCD genotype confirmation, disease severity, and specific clinical outcomes. Determining the genotype based on ICD (International Classification of Diseases) coding, in particular, has been shown to be error-prone. For this reason, we could not stratify our analysis by SCD genotype based solely on administrative data; however, we are confident that our study cohort met the case definition for SCD (any sickling hemoglobinopathy) based on a count of included ICD codes within a 5-year period. Differing genotypes may partially explain the difference between cases and controls found in this study. We do not know if individuals had a palliative care consult as we rely on administrative data and the encounter for palliative care code (Z51.5, previously V66.7) has poor sensitivity.48,49 Our study highlights the importance of better understanding specialty palliative care's role in individuals with SCD. Finally, the method of determining cases depends on inclusion in the data sources made available to the SCDC program. Those individuals who are not insured by Medicaid or other California state programs or those who did not go to the hospital or ED three or more times within any five-year period are likely to be excluded, which biases our sample toward a sicker population. Given the chronic and progressive nature of SCD, we expect this population to be small.
In conclusion, people with SCD are dying suddenly and at a young age. Comprehensive hematologic care of people with SCD needs to include high-quality acute care and advance care planning. The latter will be challenging as many of these conversations need to occur at community hospitals where adults with SCD are primarily receiving acute care and during the transition from pediatric-to-adult care, which is an already overwhelming time for young people with SCD. Therefore, many patients may require specialized palliative care integration into their SCD team so that their advance care planning needs and goal-directed care can be fully addressed. Research and policy priorities also need to focus on how to standardize high-quality acute care in emergency settings for this population and to determine best practices for integrating advance care planning conversations and decision making into the care of all people with SCD.
Footnotes
Acknowledgments
The Sickle Cell Data Collection Program is funded by the CDC Foundation through grants from Pfizer, Inc., Bioverativ, Inc., Doris Duke Charitable Foundation, and Global Blood Therapeutics.
Author Disclosure Statement
No competing financial interests exist.