
Abstract
The genetic diversity of HIV-1, driven by mutation and recombination, poses significant challenges to prevention and control efforts, particularly in regions like China where multiple subtypes and circulating recombinant forms co-circulate. Men who have sex with men (MSM) represent a key population for the emergence of novel recombinants. This study characterizes two novel unique recombinant forms (URFs) identified within the MSM population in Hebei, China. Viral RNA extraction, amplification, and near full-length genome (NFLG) sequencing were performed. Phylogenetic analysis based on NFLG alignments was conducted in MEGA 6 under the Kimura 2-parameter model with 1,000 bootstrap replicates. Recombination was assessed using the Recombinant Identification Program and SimPlot v3.5.1. Breakpoint-defined regions were phylogenetically analyzed, and recombination maps were generated. Phylogenetic and recombinant analysis based on NFLG sequences (designated BDL061 and BDL071) revealed that they originated from subtypes B and C. BDL061 exhibited a predominantly subtype B backbone with interspersed subtype C segments, while BDL071 displayed a predominantly subtype C backbone with subtype B segments. Phylogenetic analysis of recombinant segments strongly supported (bootstrap >90%) subtype B and C parental origins for the respective fragments. We report the identification and characterization of two phylogenetically distinct, novel HIV-1B/C URFs (BDL061 and BDL071) among MSM in Hebei, China. Their unique mosaic structures, differing predominant backbones, and confirmation as novel recombinants underscore the ongoing evolution and increasing complexity of the HIV-1 epidemic within this high-risk population in China. These findings highlight the critical need for NFLG-based surveillance to accurately track viral diversity and inform public health strategies.
Introduction
Since the initial report of HIV/AIDS in 1981, it has been a major global health burden threatening public health. 1 Over the past 40 years, the ongoing human immunodeficiency virus type (HIV-1) pandemic has resulted in about 40 million people currently living with HIV, accounting for a disproportionate share of the global disease burden. 2 Both mutation and recombination are key mechanisms driving the frequent evolution and genetic diversity of HIV-1. 3 These processes are primarily driven by template switching during the viral replication cycle. Furthermore, the co-circulation of distinct HIV-1 strains within a population enhances the risk of coinfection, thereby facilitating the emergence of novel recombinant forms. Circulating recombinant forms (CRFs) are defined as recombinants that exhibit a consistent recombination pattern along their near full-length genome (NFLG) and have been found in three or more epidemiologically unlinked individuals. 4 In contrast, unique recombinant forms (URFs) are recombinants that do not meet the CRF criteria. To date, 173 CRFs and numerous URFs have been reported in the Los Alamos National Laboratory (LANL) HIV database (https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html). These recombinants are particularly prevalent in regions where multiple HIV-1 subtypes and CRFs co-circulate.
Due to the large number of people living with HIV and the complex diversity of circulating subtypes, China faces significant challenges in HIV-1 prevention and control. National and regional molecular epidemiology studies have revealed CRF07_BC and CRF01_AE as the predominant subtypes, alongside an alarming increase in the diversity and complexity of HIV-1 strains circulating in China. 5 In addition, novel CRFs (e.g., CRF55_01B, CRF59_01B, CRF65_cpx) and numerous URFs, primarily comprising CRF01_AE, CRF07_BC, B and subtype C, have emerged and are increasingly reported among men who have sex with men (MSM) populations.6–8 The emergence of these novel URFs contributes significantly to the growing genetic heterogeneity of HIV-1 in China. In this study, we identified two novel URFs in Hebei, China.
Materials and Methods
Two novel HIV-1 URFs (B/C) were isolated from MSMs and designated as BDL061 and BDL071. The BDL061 strain was derived from a 51-year-old male with a plasma viral load of 1.03 × 106 copies/mL, while BDL071 originated from a 39-year-old male with a plasma viral load of 2.67 × 104 copies/mL. This study was reviewed and approved by the Institutional Medical Ethics Committee of Baoding People’s Hospital (Approval ID: 2023-04). Written informed consent was obtained from all participants prior to sample collection.
HIV-1 viral RNA extraction, amplification, and NFLG sequencing were performed according to established protocols described by Yang et al. 7 Raw sequence reads were trimmed and assembled using Sequencher v5.4.6, generating consensus NFLG sequences approximately 9.0 kb in length. Using the HIV-1 reference HXB2 (GenBank accession number: K03455) as the coordinate system, the genomic positions of the obtained sequences were mapped using the HIV Sequence Locator (https://www.hiv.lanl.gov/content/sequence/LOCATE/locate.html). The NFLG sequence of strain BDL061 spanned 8913 bp (HXB2 coordinates 765 to 9,616), while that of BDL071 spanned 8,983 bp (HXB2 coordinates 632 to 9,556). Both NFLG sequences encompassed all major structural (gag, pol, env) and regulatory (tat, rev, vif, vpr, vpu, nef) genes of HIV-1. The two NFLG sequences were aligned with HIV-1 subtype reference sequences obtained from the LANL HIV database (https://hiv.lanl.gov/components/sequence/HIV/search/search.html) using MAFFT v7.4.8. The resulting alignment was manually adjusted, and terminal regions were trimmed using BioEdit version 7.2.5.0. Subsequently, a phylogenetic tree was constructed based on the NFLG alignment in MEGA 6 using the Kimura 2-parametric model with 1000 bootstrap replicates. Potential recombination was initially screened using the online Recombinant Identification Program (RIP). Putative recombination breakpoints were then determined and verified using SimPlot v 3.5.1. Phylogenetic trees for individual genomic regions defined by the breakpoints were constructed using the same methodology described for the NFLG tree. Finally, a schematic genomic map illustrating the recombination structure of the novel HIV-1 recombinant was generated using the online Recombinant HIV-1 Drawing Tool (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html).
Results
Phylogenetic analysis based on NFLG revealed that the BDL061 and BDL071 sequences each formed a distinct monophyletic branch, separate from established subtypes and CRFs (Fig. 1). Within the tree, the BDL061 NFLG sequence clustered closest to CRF107_01B, while BDL071 clustered closest to CRF57_BC reference sequences. To confirm their novelty, the BDL061 and BDL071 NFLG sequences were queried against the LANL HIV database using the HIV BLAST tool (https://www.hiv.lanl.gov/content/sequence/BASIC_BLAST/basic_blast.html). No sequences exhibiting >95% similarity across the NFLG were identified, supporting their classification as novel URFs.Recombination breakpoints within the NFLG of BDL061 and BDL071 were determined using SimPlot (v3.5.1) and the RIP (https://www.hiv.lanl.gov/content/sequence/RIP/RIP.html). Analysis confirmed both sequences are inter-subtype B/C recombinants. Notably, the predominant genomic backbone of BDL061 was subtype B, whereas subtype C constituted the predominant backbone in BDL071. Schematic recombination maps were generated using the Recombinant HIV-1 Drawing Tool (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html) based on HXB2 coordinates (Figs. 2 and 3).The recombinant mosaic structures are defined as follows: IC (HXB2, 790-1287nt); IIB (HXB2, 1288-5909nt); IIIC (HXB2, 5910-6198nt); IVB (HXB2, 6198-9616nt), BDL061; IC (HXB2, 790-1278nt); IIB (HXB2, 1279-2143nt); IIIC (HXB2, 2144-9556nt), BDL071.

Phylogenetic tree analysis. The neighbor-joining phylogenetic tree of NFLG sequences of BDL061 () and BDL071 ( ) was constructed using Mega6.0. The stability of each node was assessed by bootstrap tests with 1,000 replicates, and only bootstrap values ≥80% were shown at the corresponding nodes. The scale bar represents 5% genetic distance. NFLG, near full-length genome.
) was constructed using Mega6.0. The stability of each node was assessed by bootstrap tests with 1,000 replicates, and only bootstrap values ≥80% were shown at the corresponding nodes. The scale bar represents 5% genetic distance. NFLG, near full-length genome.

Recombination breakpoints analysis of the NFLG sequence of BDL061 and BDL071.
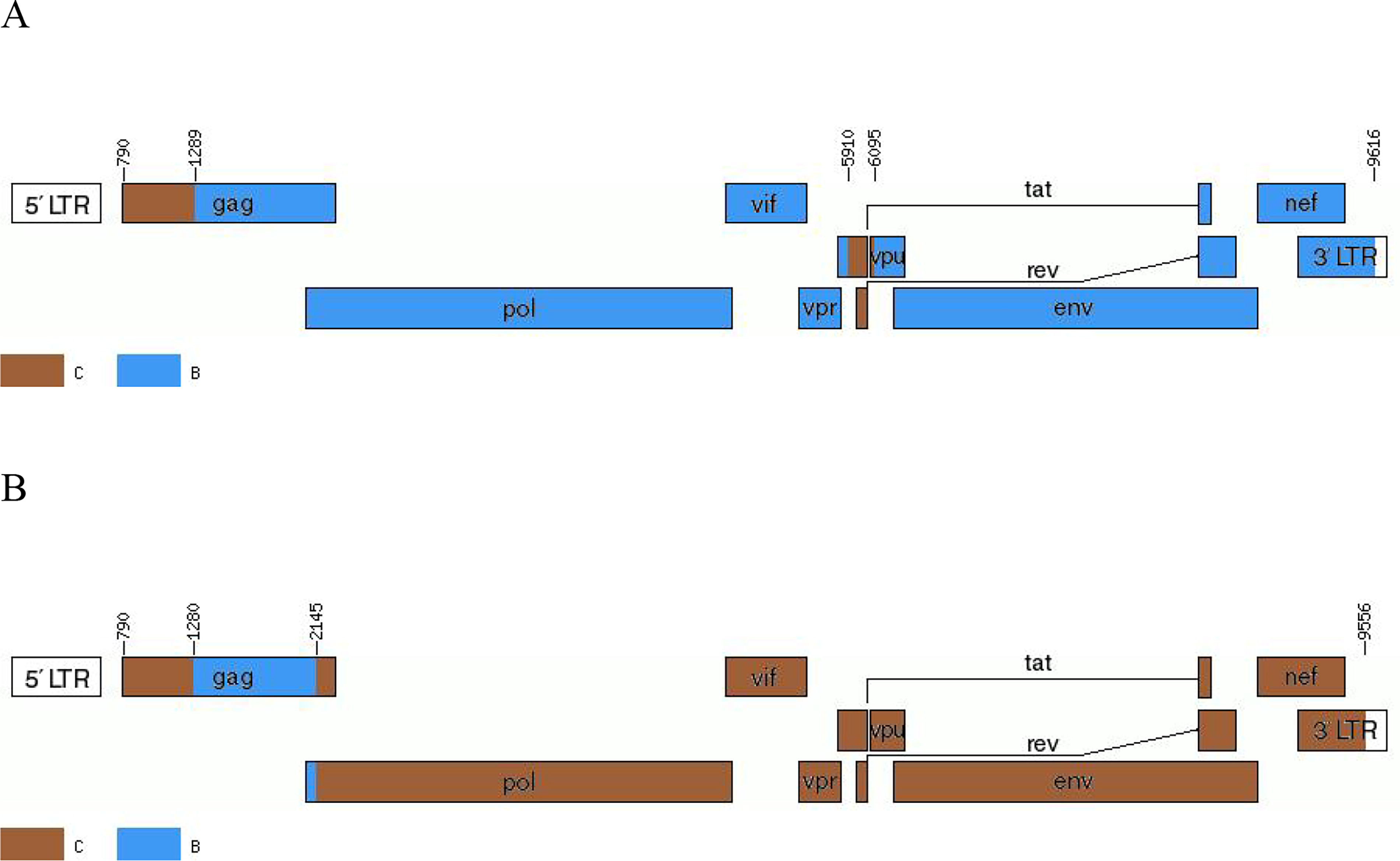
Genetic maps of BDL061
Phylogenetic trees were constructed for each recombinant segment defined by the breakpoints. Analysis demonstrated strong statistical support (bootstrap value >90%) for the subtype B segments clustering with subtype B references and the subtype C segments clustering with subtype C references (Fig. 4), confirming the parental origins of the recombinant fragments.
Subregion phylogenetic tree. Subregion phylogenetic analysis of different segments of BDL061 ) marks BDL061 and BDL071. Bootstrap values ≥90% were shown at the corresponding nodes. The scale bar represents 5% genetic distance.
Discussion
In this study, we characterize two novel and phylogenetically distinct HIV-1B/C recombinant forms (designated BDL061 and BDL071) identified within MSM populations in Hebei province, Northern China. Hebei represents a critical region where the HIV epidemic is predominantly fueled by MSM 9 and has recently emerged as a hotspot for the genesis of novel CRFs and URFs.10–11 This burgeoning genetic diversity creates a fertile landscape for viral recombination and evolution of HIV-1.
Crucially, the recombination architecture of BDL061 and BDL071 deviates significantly from other URFs reported in China. While the majority of reported URFs stem from recombination between CRF01_AE and CRF07_BC lineages,12–14 our findings document the emergence of inter-subtype B/C recombination within the MSM population. A defining feature of the two novel URFs is the localization of key recombination breakpoints within gag region—a genomic segment not routinely targeted by standard subtyping protocols. This observation underscores a critical methodological limitation: the widespread reliance on partial pol sequences for HIV-1 subtyping15–16 is demonstrably inadequate for detecting complex recombinants, particularly those involving breakpoints outside pol. Such limitations inevitably lead to misclassification and an incomplete understanding of true viral diversity. Consequently, our findings compellingly argue for the adoption of NFLG or full-length genomic sequencing as the essential standard for robust identification and characterization of emerging recombinant strains, especially within dynamic transmission networks like MSM.
The continuous expansion of HIV-1 genetic diversity and the recurrent emergence of novel recombinants, exemplified by these distinct B/C URFs, demand heightened vigilance. The detection of recombinants with parental lineages (B and C) and breakpoint patterns divergent from the dominant CRF01_AE/CRF07_BC paradigm signals evolving complexities within the MSM transmission networks in Hebei. It highlights the dynamic molecular evolution of HIV-1 and underscores the urgent need to leverage comprehensive genetic surveillance data to refine real-time epidemiological tracking and inform targeted prevention strategies aimed at curbing the spread of HIV-1 in this and similar settings.
Sequences Data
The gene sequences of BDL061 and BDL071 were deposited in the GenBank with the accession numbers PV567363and PV567364, respectively.
Authors’ Contributions
X.Z. and Z.C.: Article writing, experimental operation, data analysis. J.D.: RNA extraction and sequence assembly. H.S.: Sample information collection and sorting. S.C.: Conducting HIV confirmatory tests. W.F.: Experimental design and article correction.
Footnotes
Author Disclosure Statement
No competing financial interest exists.
Funding Information
This study was supported by the Medical Scientific Research Project of Hebei (No: 20240874).