
Abstract
In China, the HIV-1 epidemic is predominantly dominated by the recombinant strains CRF01_AE and CRF07_BC. The high replication rate, error-prone reverse transcriptase, and frequent recombination events of HIV-1 have facilitated the emergence of circulating recombinant forms (CRFs) between these major lineages. In this study, a novel HIV-1 second-generation circulating recombinant form (CRF193_0107), consisting of CRF01_AE and CRF07_BC, was identified during routine molecular surveillance in Hebei Province, China. We successfully obtained near-full-length genome (NFLG) sequences from three samples with no epidemiological link and performed phylogenetic, recombination, and temporal evolutionary analyses. Phylogenetic analysis revealed that these NFLG sequences formed a distinct monophyletic cluster with a high bootstrap value. Recombination analysis showed that all NFLGs shared the same unique mosaic recombination pattern between CRF01_AE and CRF07_BC clades, with one CRF07_BC fragment in the vif-vpr-tat region (HXB2 positions 5,550–5,965) inserted into a CRF01_AE backbone. Further subregion phylogenetic analysis confirmed that segment I+III of CRF193_0107 originated from men who have sex with men (MSM)-associated CRF01_AE cluster 4, and segment Ⅱ from MSM-associated CRF07_BC cluster N. The temporal evolution analysis indicated that CRF193_0107 originated in 2016. The emergence of CRF193_0107 underscores the importance of monitoring HIV-1 second-generation recombinant forms (CRFs_0107), particularly the transmission and evolution among men who have sex with men (MSM).
Keywords
Over the past few decades, CRF07_BC and CRF01_AE have become the predominant circulating HIV-1 strains in China. These two subtypes collectively account for over 50% of cases in most regions nationwide, exhibiting particularly dominant prevalence among men who have sex with men (MSM). 1 When these two predominant subtypes co-circulate in the same region, high-risk behaviors among MSM populations may increase the opportunity of dual infection with both CRF01_AE and CRF07_BC, as well as the emergence of second-generation recombinant strains. Viral evolution through recombination facilitates enhanced biological fitness and evasion of host immune responses, thereby complicating diagnosis, clinical management, and vaccine development against these subtypes. 2 Consistent with this, recent reports have highlighted the continuous emergence of second-generation CRFs (composed of CRF01_AE and CRF07_BC) among MSM in China.3,4 According to the latest documentation, at least 21 such CRFs_0107 have been cataloged in the HIV database of Los Alamos National Laboratory. 5 In this study, a novel emerging second-generation circulating recombinant form (designated CRF193_0107) was identified among MSM in Hebei province, China. We characterized its genetic characterization, recombination pattern, and evolutionary history.
During continuous surveillance of HIV-1 molecular epidemiology and pretreatment drug resistance in Hebei Province, China, from our previous study, a monophyletic transmission cluster comprising three treatment-naive HIV-1-positive MSM was identified through near-full-length genome (NFLG) quasispecies sequencing. All participants provided written informed consent prior to specimen collection and completed standardized epidemiological questionnaires. No direct epidemiological linkages were established among these cases. Their baseline demographic characteristic profiles are summarized in Table 1.
Demographic Characteristics of Participants Infected with HIV-1 CRF193_0107

MSM, men who have sex with men.
Patients HB010038, HB010280, and HB010298 were diagnosed in 2020, 2021, and 2021, respectively. Viral RNA was extracted from plasma using the QIAamp Viral RNA Mini Kit (Qiagen). Three-fragment reverse transcription was performed using specific primers and the SuperScript™ IV First-Strand Synthesis System (Invitrogen) to generate cDNA covering the NFLG. Viral quasispecies sequence DNA products were obtained through high-fidelity nested PCR amplification targeting the gag, pol, and 3'-half regions.6,7 For the second round of amplification, specific primers with dual-end barcode tags were utilized. Sequencing was conducted on the PacBio Revio platform. Raw sequencing data were demultiplexed and filtered. Subsequently, a bioinformatics pipeline was employed for quasispecies sequence extraction. For each fragment from the same infected individual, the top 10 most abundant quasispecies sequences were included in this study. These sequences have been submitted to GenBank with accession numbers PV743059-PV743140.
The phylogenetic analysis revealed that the consensus sequence of the top 10 quasispecies from each participant was closely related, forming a distinct monophyletic clade (bootstrap value = 1) among all known CRFs circulating in China (Fig. 1). This suggests that they originated from a single ancestor and might represent a potential novel CRF. Further recombination analysis of the recombinant identification program indicated that they shared a common recombinant breakpoint structure pattern (Fig. 2). Bootscan and jpHMM analysis further confirmed that these sequences were composed of CRF01_AE and CRF07_BC, delineated into three segments by two breakpoints (CRF01_AE subregions: I+III, 8407 bp; CRF07_BC subregion: Ⅱ, 416 bp) (Fig. 3A and B). Subregion phylogenetic analyses further revealed that their parental lineages were CRF01_AE cluster 4 and CRF07_BC cluster N, both of which are prevalent among MSM populations in China (Fig. 4). In summary, these strains exhibited identical recombination patterns and were distinct from any previously reported CRFs. Importantly, the three participants infected with these strains had no direct epidemiological linkages. Therefore, according to the standardized HIV nomenclature principles of the HIV database, these strains are designated as a novel CRF, named CRF193_0107.
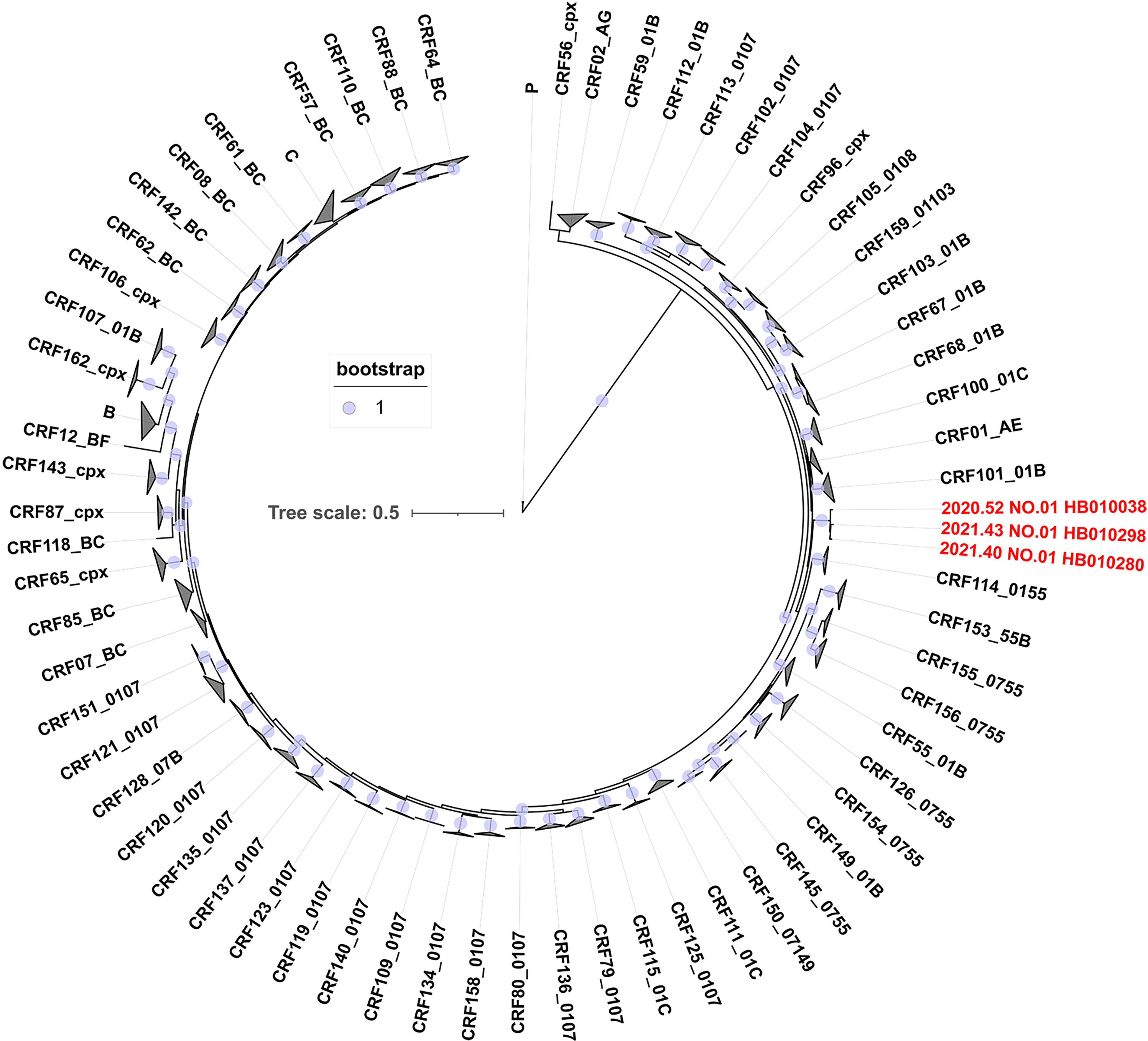
Phylogenetic analysis based on near full-length HIV-1 genome sequences of HIV-1 CRF193_0107. The maximum likelihood phylogenetic tree (PhyML) was constructed using the reference sequences of the different HIV-1 CRFs identified in China. Bootstrap = 1.0 is indicated by solid circle. The sequences of CRF193_0107 are highlighted in red.
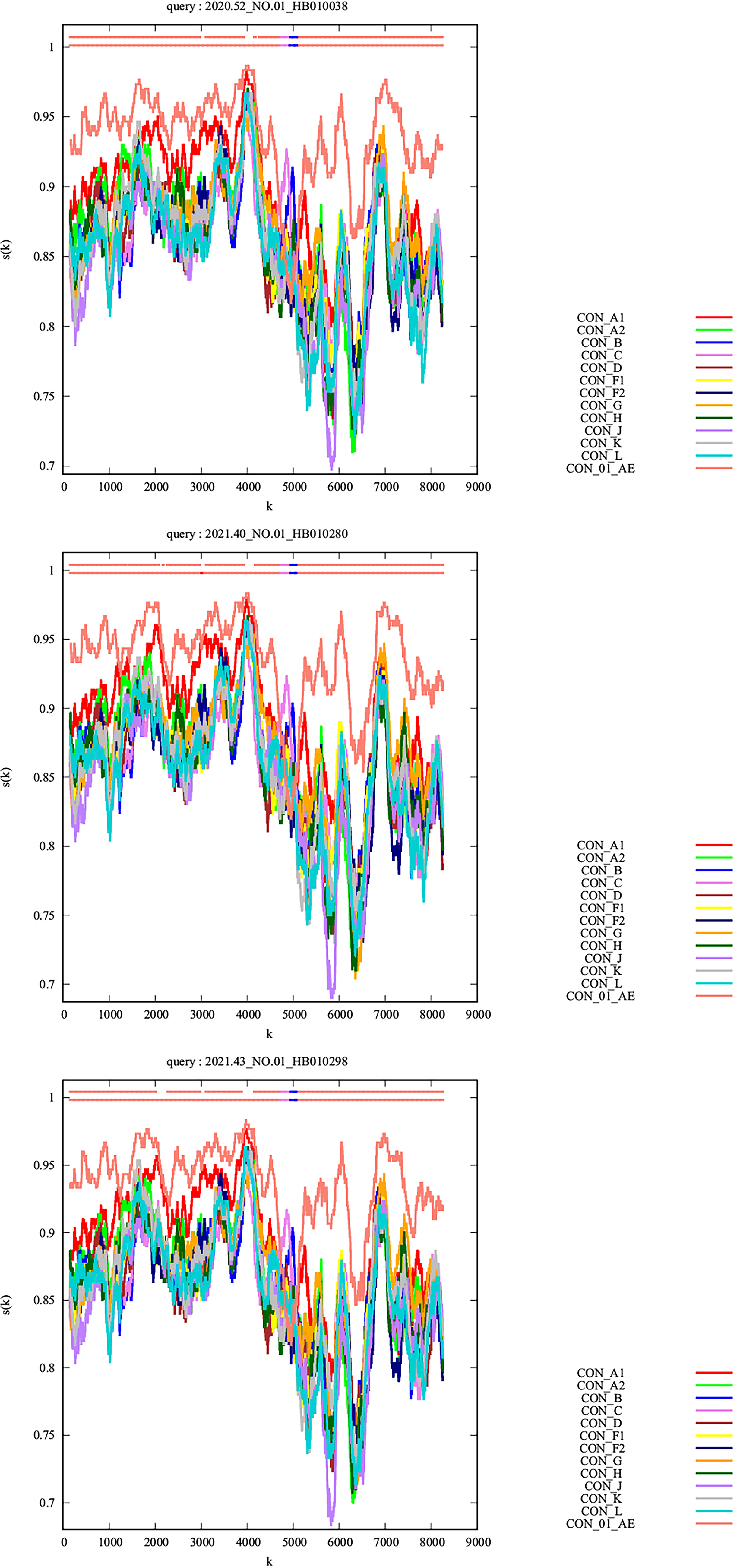
Recombinant Identification Program analysis based on near full-length HIV-1 genome sequences of HIV-1 CRF193_0107. RIP analysis was conducted using a 400 bp window size with reference sequences of subtypes A–L + CRF01_AE.

Bootscan analysis and recombination pattern of HIV-1 CRF193_0107.
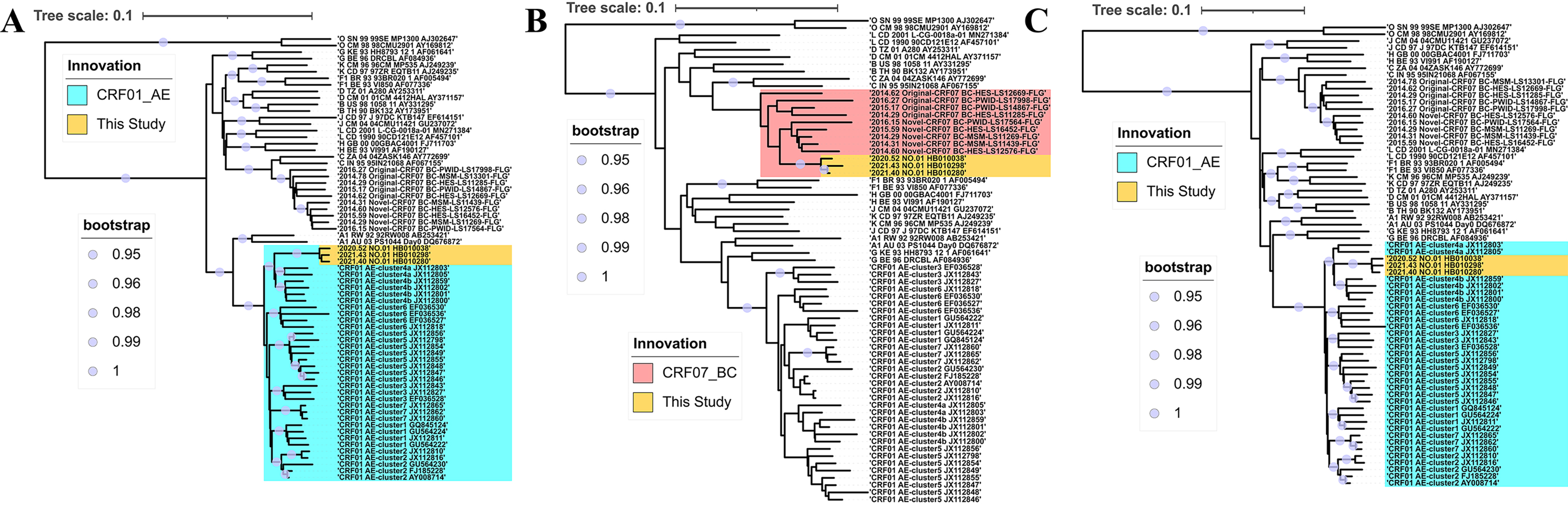
Subregion tree of HIV-1 CRF193_0107. The subregion trees of the three mosaic fragments identified by recombinant analysis were constructed using the neighbor-joining method based on the Kimura 2-parameter model in MEGA. Bootstrap = 0.95–1.0 is indicated by solid circle. The included reference sequences include group O, 10 subtypes of group M, clusters 1–7 of CRF01_AE, and clusters N and O of CRF07_BC.
To trace the origin of CRF193_0107, we conducted a temporal evolutionary analysis using the Bayesian molecular clock method implemented in BEAST version 1.10.4. 8 For this analysis, we selected the top 10 viral quasispecies sequences from each infected individual to compile our dataset to obtain more reliable estimate. Subsequently, we performed Bayesian evolutionary analyses on the CRF01_AE subregion (Igag), CRF01_AE subregion (Ipol), CRF07_BC subregion (II), and the combined CRF01_AE subregion (Ivif+III) to estimate the time to the most recent common ancestor (tMRCA). As shown in Figure 5, the results indicated that the tMRCA of the four subregions of CRF193_0107, with 95% highest probability density (HPD), were 2016.84 (95% HPD: 2015.61–2017.81), 2016.36 (95% HPD: 2015.13–2017.37), 2016.50 (95% HPD: 2014.43–2018.33), and 2016.50 (95% HPD: 2015.27–2017.63), respectively. These findings suggest that the tMRCA of the CRF193_0107 strain likely emerged around 2016.
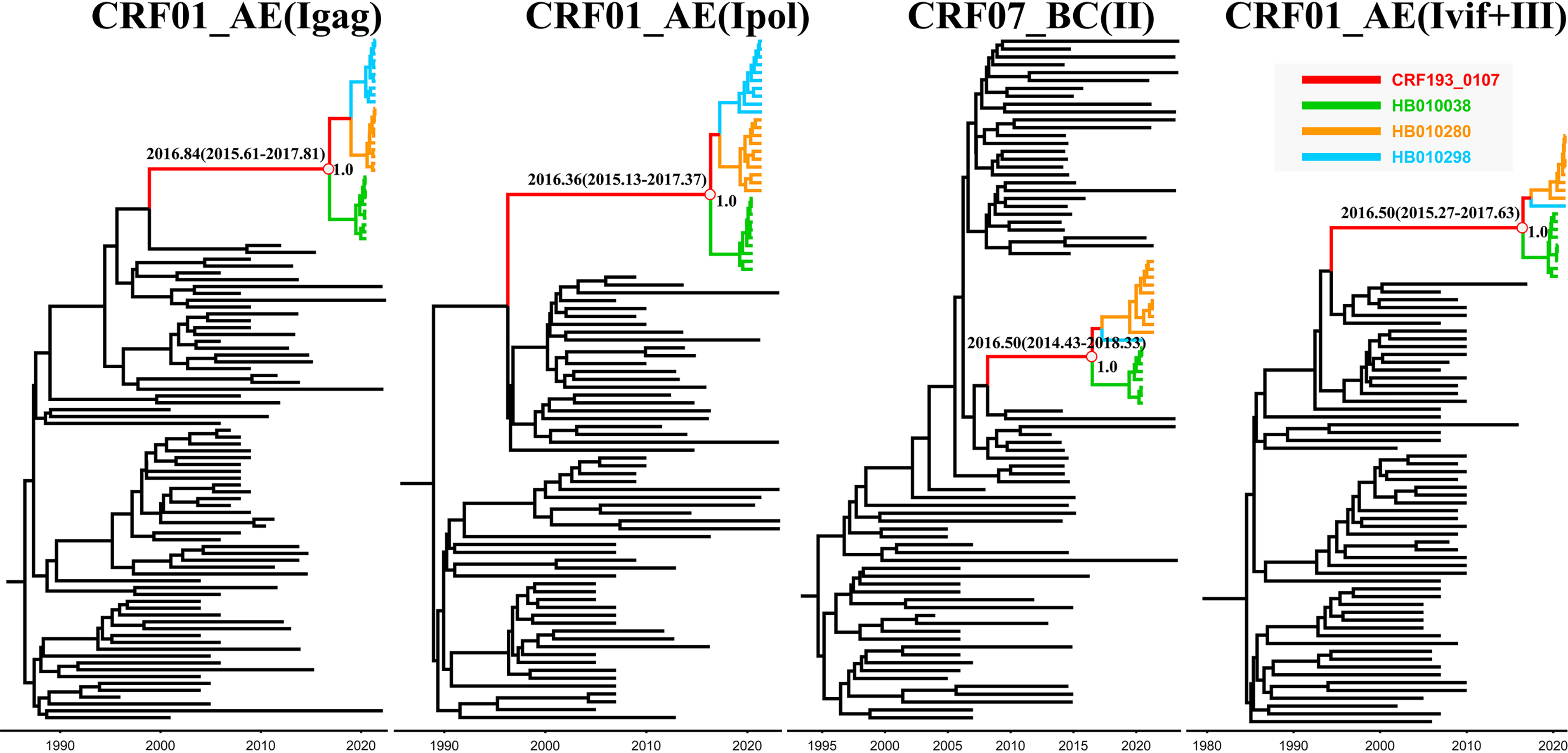
Evolutionary analysis of subregions from CRF193_0107. The maximum clade credibility (MCC) trees of CRF01_AE segments (Igag), CRF01_AE segments (Ipol), CRF07_BC segments (Ⅱ), and combined CRF01_AE segments (Ivif+III), respectively. The posteriors, the mean tMRCA, and the 95% HPD for the key nodes are indicated.
Recently, the proportion of HIV-1 infections among MSM in Hebei Province, China, has seen an increase, with CRF01_AE and CRF07_BC emerging as the predominant subtypes of HIV-1 in this region. The potential for dual infections with CRF01_AE and CRF07_BC strains may increase, providing more opportunities for the generation of novel supergroups such as CRF01_AE/CRF07_BC in Hebei Province, China.9,10 Previously, CRF123_0107 and CRF140_0107 have been identified among MSM in this area.5,11 Notably, CRF123_0107, CRF140_0107, and CRF193_0107 share a common genomic structural feature, namely, the pol gene fragment region (HXB2: 2253–3870) belonging purely to the CRF01_AE subtype. This characteristic makes these strains highly susceptible to being overlooked in drug resistance monitoring and misclassified as parental strains, leading to their concealed prevalence. Consequently, due to their ability to “evade detection,” strains originating from the MSM population, such as CRF193_0107, may circulate more widely and pose a greater threat to public health. Undoubtedly, this poses challenges for disease surveillance and contributes to an increased disease burden.
In summary, we have identified a novel CRF (CRF193_0107) among MSM in Hebei Province, with its origin traced back to approximately 2016. Although Hebei is classified as a low HIV prevalence region in China, the identification of this novel CRF among the MSM population, the key group of the local epidemic, suggests an increasing genetic diversity of HIV-1 in this area and a more complex epidemic dynamic than previously recognized. Therefore, further molecular surveillance of HIV-1 recombinant strains, particularly those originating from CRF01_AE and CRF07_BC, among MSM in this region is essential. Such efforts are critically important for monitoring local viral evolution and informing public health intervention strategies.
Authors’ Contributions
B.Z., J.H., W.M., and L.L. designed the study. B.Z. performed the experiments. B.Z. and X.R.W. participated in data analyses. All authors participated in the writing process, the article correction, and approved the final article.
Footnotes
Acknowledgments
The authors thank the members of the Fifth Hospital of Shijiazhuang for the collection of samples and epidemiological data, and thank all of the participants and peer workers.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (82173583), the State Key Laboratory of Pathogen and Biosecurity (AMMS).
Sequences Data
The gene sequences were deposited in GenBank with the accession numbers PV743059-PV743140.