
Abstract
Mutation and recombination are key mechanisms in the adaptation, spread, and genetic diversity of human immunodeficiency virus (HIV)-1. We identified two novel unique recombinant forms (URFs) from two persons living with HIV-1, both infected through homosexual contact in Hebei, China. Phylogenetic and recombinant analysis based on the near full-length genome (NFLG) sequences revealed that they originated from CRF01_AE and B. NFLG sequencing revealed complex recombinant patterns in both samples, indicating ongoing recombination and co-infection in men who have sex with men (MSM). The emergence of novel URFs underscores the complexity of the HIV-1 epidemic among MSM and highlights its evolving molecular diversity and epidemiological implications.
Keywords
Introduction
Since the initial report of human immunodeficiency virus (HIV) in 1981, the acquired immunodeficiency syndrome (AIDS) pandemic remains a major global health burden. An estimated 40.8 million people globally were living with HIV as of 2024, with 1.3 million people newly acquiring HIV and 630,000 people dying from AIDS-related illnesses. 1 Although significant progress has been made in scaling up antiretroviral therapy, reaching 77% coverage among people living with HIV, 2 the ambitious target to end AIDS as a public health threat by 2030 faces persistent obstacles, among which the remarkable genetic diversity and continuous evolution of HIV-1 are paramount.
The extensive genetic variability of HIV-1 is fueled primarily by its error-prone reverse transcription and frequent recombination events. 1 Co-infection with distinct viral strains can lead to template switching during replication, generating unique recombinant forms (URFs), which may establish ongoing transmission. Upon identification in three or more epidemiologically unlinked individuals, URFs are designated as circulating recombinant forms (CRFs). 3 To date, the Los Alamos National Laboratory (LANL) HIV database (https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html) has documented 173 CRFs alongside numerous URFs, underscoring recombination as a key driver of viral diversity, particularly in regions with co-circulating subtypes.
China faces significant challenges in the prevention and control of HIV-1 because of the number of persons living with HIV and complex subtype diversity. National and regional molecular epidemiology shows that the predominant subtypes are CRF07_BC and CRF01_AE, accompanied by a rapid increase in subtype diversity and complexity. 4 In addition, new CRFs and URFs, such as CRF55_01B, have emerged among men who have sex with men (MSM),5–7 contributing to the increasing genetic heterogeneity of HIV-1 in China.
In this study, two novel HIV-1 URFs (CRF01_AE and B) were isolated from MSM in Hebei Province, designated as BDL128 and BDL150, respectively. The BDL128 sample was obtained from a 33-year-old male with a viral load of 1.55 × 105 copies/mL and an initial CD4+ T cell count of 239 cells/µL; the BDL150 sample was from a 55-year-old male with a viral load of 1.54 × 105 copies/mL and an initial CD4+ T cell count of 578 cells/µL. This study was reviewed and approved by the Medical Ethics Committee of Baoding People’s Hospital (Approval ID: 2025-03). Written informed consent was obtained from participants before sample collection.
HIV-1 viral RNA extraction, amplification, and sequencing were carried out as previously described. 8 Sequence trimming and assembly were performed using Sequencher v5.4.6, providing an HIV-1 near full-length genome (NFLG) sequence of approximately 9.0 kb. Based on the reference HXB2 sequence (GenBank ID: K03455), we used the HIV Sequence Locator (https://www.hiv.lanl.gov/content/sequence/LOCATE/locate.html) to determine the relative positions of the obtained HIV-1 sequences. The NFLG sequence of BDLL128 was 8,895 bp (from nucleotide positions 777 to 9,636), while that of BDL150 was 8,854 bp (from nucleotide positions 773 to 9,626). Both NFLG sequences contained all structural and regulatory genes. The two NFLG sequences were aligned with HIV-1 standard reference sequences from the LANL HIV database (https://hiv.lanl.gov/components/sequence/HIV/search/search.html) using MAFFT v6. Manual adjustment and terminal sequence trimming were performed in BioEdit v7.2.5.0. A phylogenetic tree based on the NFLG sequences was constructed in MEGA 6 with 1,000 bootstrap replicates under the Kimura 2-parameter model.
BDL128 and BDL150 clustered with subtype B strains in the phylogenetic analysis and were distinctly separated from other subtypes, forming a distinct monophyletic cluster (Fig. 1). The HIV BLAST (https://www.hiv.lanl.gov/content/sequence/BASIC_BLAST/basic_blast.html) database failed to retrieve any sequences with >95% similarity, indicating that the two sequences are likely novel URFs.
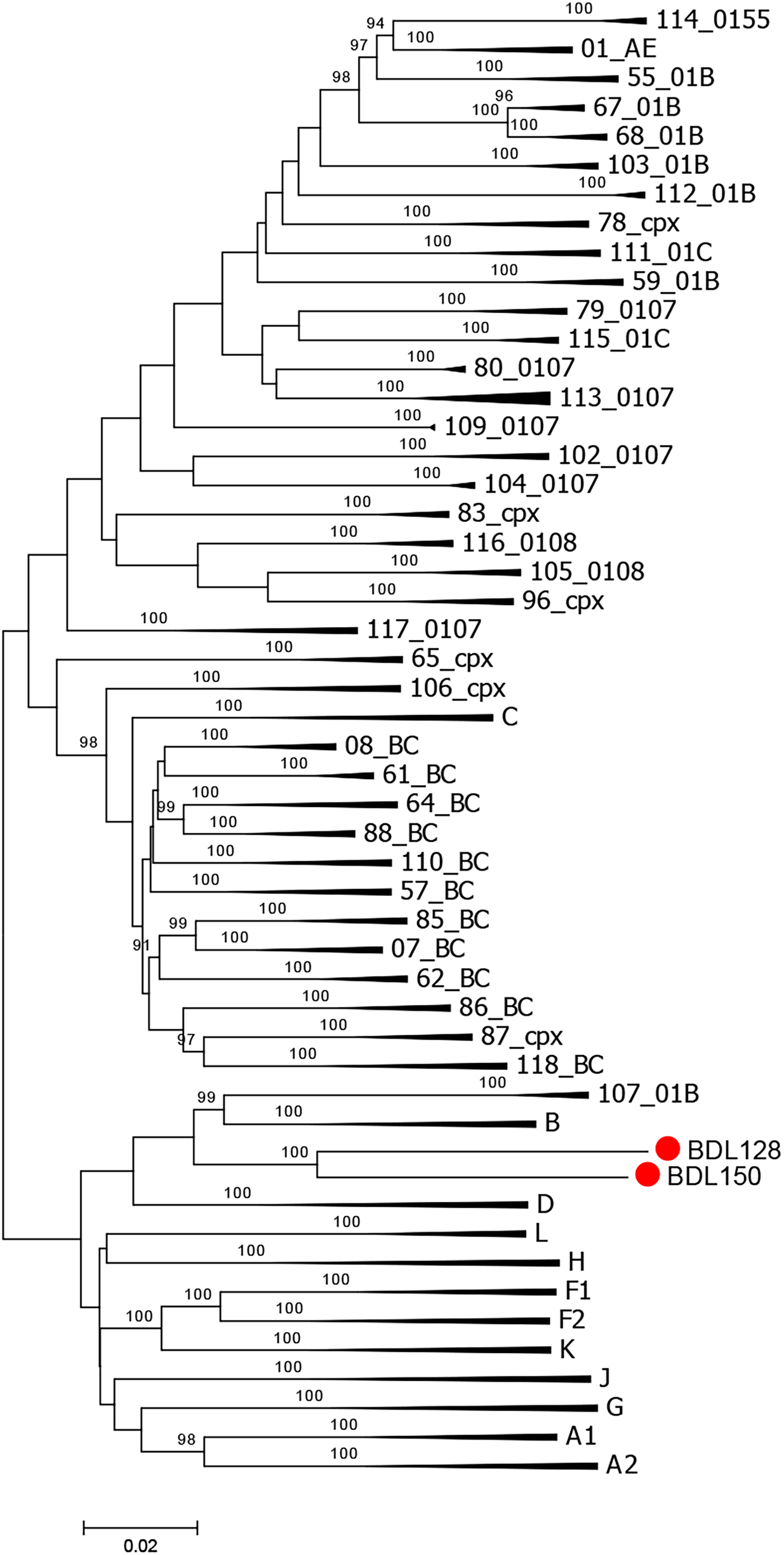
Phylogenetic tree analysis. The neighbor-joining phylogenetic tree of BDL128 and BDL150 was constructed using Mega v6 based on NFLG sequences. The stability of each node was assessed by bootstrap tests with 1,000 replicates, and only bootstrap values ≥90% were shown at the corresponding nodes. The scale bar represents 5% genetic distance. NFGL, near full-length genome.
Recombination breakpoints of the two NFLG sequences were identified using SimPlot v3.5.1 and the Recombination Identification Program (https://hiv.lanl.gov/contens/sequence/RIP/RIP.html). Both sequences consisted of CRF01_AE and B subtypes, with the B subtype serving as the predominant genomic backbone. The breakpoints were submitted to the Recombinant HIV-1 Drawing Tool (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html) to better visualize the recombination map, and the recombination patterns were redrawn based on the HXB2 reference sequence (Figs. 2 and 3). The recombinant mosaic structure of the two sequences was described as follows: ICRF01_AE (HXB2, 790-2138nt); IIB (HXB2, 2139-6525nt); IIICRF01_AE (HXB2, 6526-7579nt); IVB (HXB2, 7580-9401nt), BDL128; IB (HXB2, 790-1183nt); IICRF01_AE (HXB2, 1184-1862nt); IIIB (HXB2, 1863-6185nt), IVCRF01_AE (HXB2, 6186-8273nt), IVB (HXB2, 8274-9408nt), BDL150.
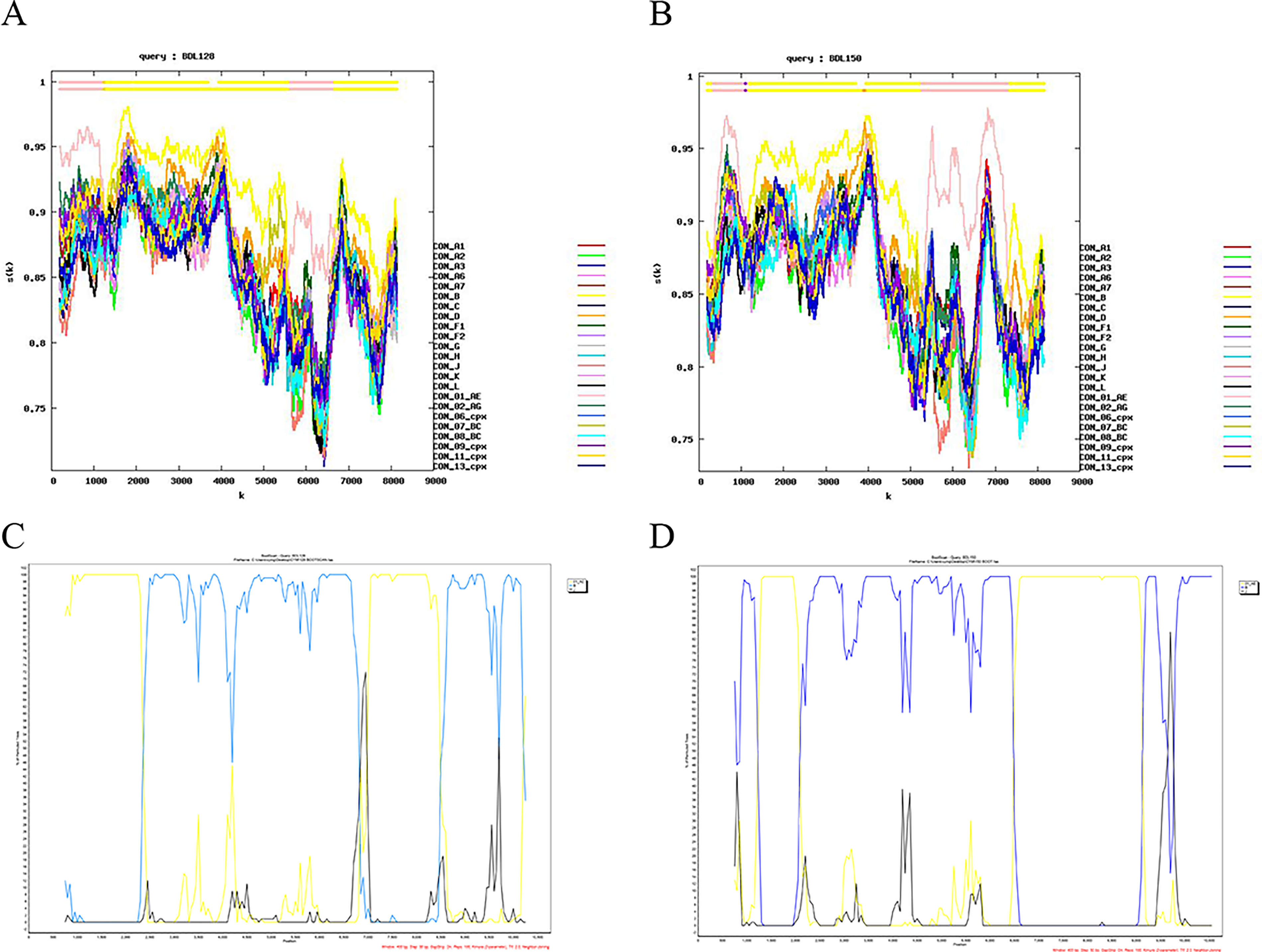
Recombination breakpoint analysis of the NFLG sequence of BDL128 and BDL150.
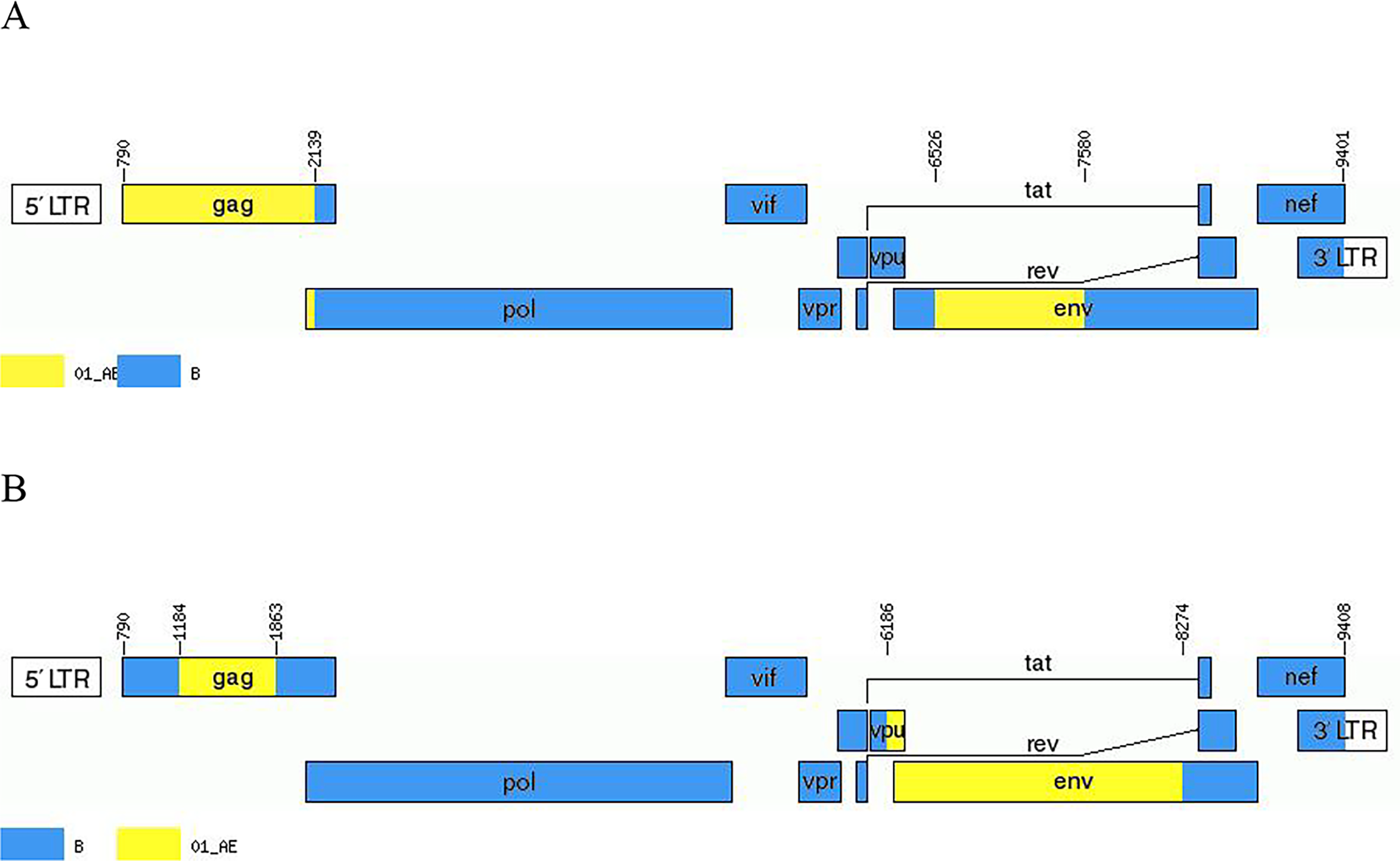
Genetic maps of BDL128
Phylogenetic trees based on the recombinant segments were constructed using MEGA v6. The parental origin of the subtype B segments in the two NFLG sequences was closely related to subtype B strains (Fig. 4). In addition, CRF01_AE segments of BDL128 formed a well-supported monophyletic cluster with CRF01_AE Cluster 4 sequences, which predominantly circulate in MSM; CRF01_AE fragments from BDL150 clustered with CRF01_AE Cluster 5, which predominantly circulates in both MSM and heterosexual populations. Combined with the epidemiological data (no reported travel history), these URFs most likely arose from in vivo recombination between locally co-circulating CRF01_AE and subtype B strains, rather than representing direct imports. However, this does not preclude the possibility that such recombinants could gain a transmission advantage and eventually evolve into new local CRFs.
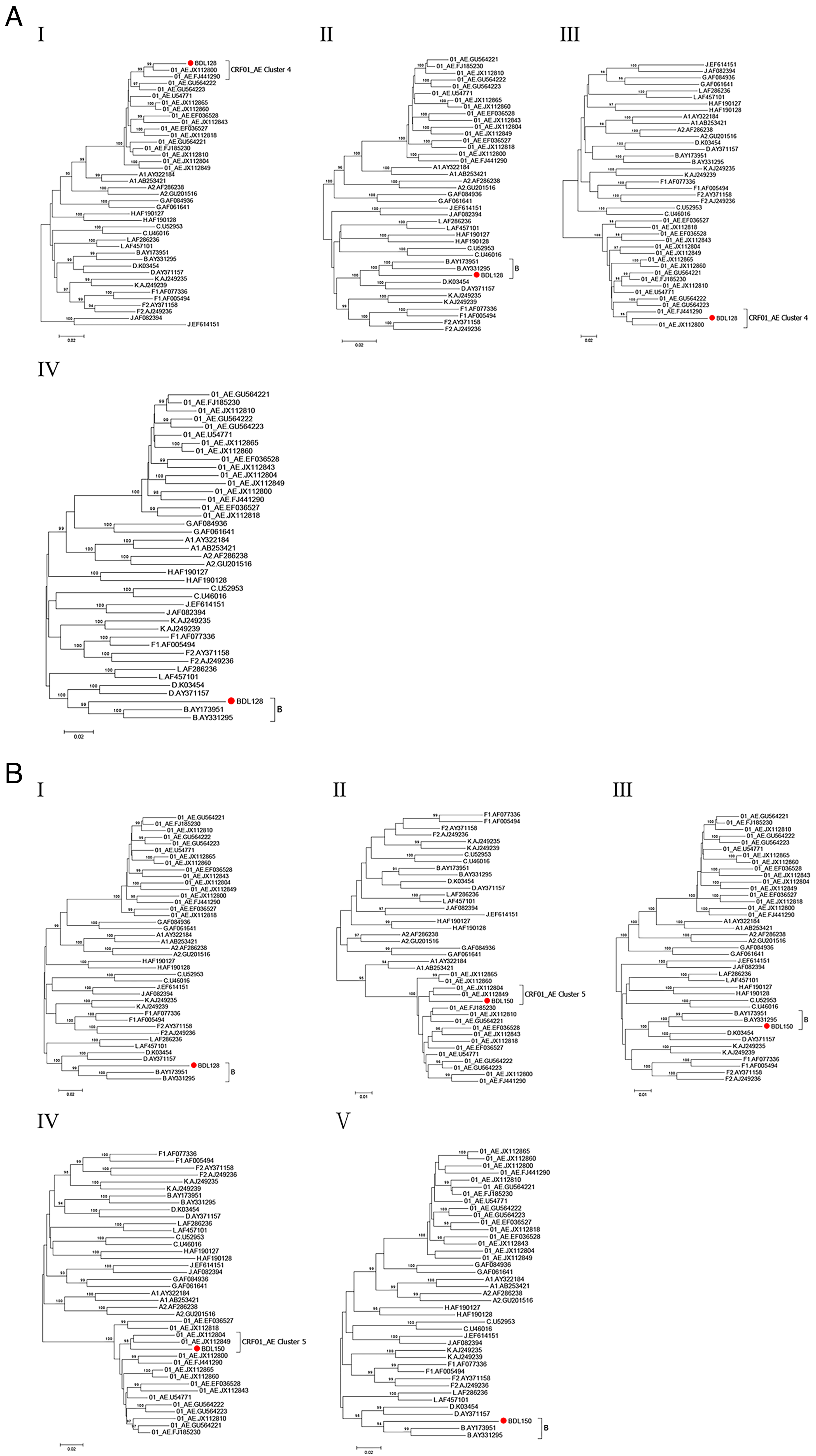
Subregion phylogenetic tree. Subregion phylogenetic analyses of different segments of BDL128
In summary, we report two novel CRF01_AE/B recombinant forms from MSM in Hebei province, located in northern China. Hebei Province is one of the provinces where the HIV epidemic is driven by MSM. 9 Recent reports of new CRFs and URFs in this region highlight it as a hotspot for HIV-1 genetic recombination and diversification,10,11 warranting close molecular surveillance.
Unlike the majority of previously reported URFs in China, which predominantly involve recombination between CRF01_AE and CRF07_BC,12–14 the recombination patterns of BDL128 and BDL150 involved CRF01_AE and subtype B, with breakpoints located in the gag and env regions. This underscores a significant limitation of routine surveillance strategies based on partial pol gene sequencing, which may fail to capture recombination events in other parts of the HIV-1 genome, underestimating local genetic complexity. Our findings align with a recent study from Guangdong, 15 where an F2/CRF02_AG recombinant would have been missed by pol-only analysis, highlighting the necessity to establish a more practical method for the detection and identification of recombinant HIV strains.
In conclusion, the discovery of novel CRF01_AE/B URFs among MSM in Hebei Province is a significant marker of the ongoing evolution and adaptation of HIV-1 in China. Available data should be utilized to enhance surveillance and prevention efforts against HIV-1 spread. Future studies should aim to expand the temporal and geographical scope of such surveillance, employing deep sequencing to gain a more comprehensive understanding of the dynamics, drivers, and long-term implications of HIV-1 recombination events on the epidemic trajectory.
Data Availability
BDL128 and BDL150 sequences were deposited in GenBank with the accession numbers PX970455 and PX970456, respectively.
Footnotes
Acknowledgments
Author Disclosure Statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding Information
This study was supported by the Baoding Science and Technology Plan Project (No. 2541ZF086).