
Abstract
Background
The alternative splicing (AS) of MAPT, which encodes Tau, in the adult human brain produces six major isoforms that play critical roles in the pathogenesis of tauopathies including Alzheimer's disease. Previous efforts have failed to differentiate human induced pluripotent stem cells (hiPSCs) to cortical neurons expressing the six isoforms of Tau.
Objective
We aim to develop a differentiation method capable of producing the six Tau isoforms in hiPSC-derived cortical neurons.
Methods
We searched for the optimal concentration, duration and treatment window of morphogens in the differentiation of hiPSCs through embryoid bodies (EBs) to dorsal forebrain neuroepithelial cells then to cortical neurons.
Results
The combined inhibition of WNT, SHH, and SMAD signaling in EBs generated neuroepithelial cells expressing appropriate dorsal forebrain markers, while suppressing ventral, midbrain, and hindbrain genes. Further differentiation in neurogenic and neurotrophic factors produced MAP2+ neurons at day 18. The iPSC-derived neurons expressed markers of all cortical layers and exhibited synapse formation and synaptic physiology. In addition, MAP2+ neurons and mitotic cells expressing radial glial markers formed aggregates that could be dissociated to produce mature neurons with similar properties. Most importantly, the six Tau isoforms were expressed from day 80 in a developmentally regulated manner, modeling the situation in human brains on an accelerated timeline.
Conclusions
This chemically defined differentiation method produces a key hallmark of mature human cortical neurons by expressing the six main splicing isoforms of Tau. It will greatly facilitate disease modeling and therapeutic discovery for many human brain disorders involving cortical neurons.
Keywords
Introduction
The human cerebral cortex plays essential roles in many high-level brain functions including cognition, emotion, memory, and perception. 1 Dysfunction of cortical neurons underlies the etiology of many neurological and neuropsychiatric disorders, such as Alzheimer's disease, Lewy body dementia, autism, depression, and schizophrenia. 2 The dramatic expansion of the human cortex, in comparison to that of the mouse, produces many human-specific features and diseases that are difficult to model in mice. 3 One prominent example is tauopathies, a group of neurodegenerative disorders with intracellular Tau inclusions induced by mutations or the abnormal alternative splicing of the MAPT gene, which encodes the microtubule-binding protein Tau. 4 In adult human brains, six main splicing isoforms of Tau are produced, with three possible combinations in the N-terminus [0N, 1N (including exon 2), or 2N (including exons 2 and 3)] and two possible combinations in the microtubule-binding domain [3R, and 4R (including exon 10)].5,6 Human fetal brains mainly produce 0N3R Tau, which does not include alternative exons 2, 3, and 10. The AS of MAPT is regulated differently in the mouse brain, 6 which has stimulated substantial efforts to generate human cortical neurons expressing Tau AS isoforms similar to that in adult human brains, in particular, the approximately equal ratio of 3R and 4R Tau. 7 Previous studies have shown that hiPSC-derived cortical neurons express only 3R Tau, with barely any 4R Tau,8–11 even after 1 year of differentiation. 8 The AS of exons 2 and 3 has not been examined in hiPSC-derived neurons. These difficulties prompted the development of microRNA-induced neurons directly converted from skin fibroblasts 12 and NGN2-induced neurons derived from iPSCs with gene-edited 4R alleles 13 to study mixed 3R/4R and 4R tauopathies.
These obstacles show that the currently available methods to differentiate hiPSCs to cortical neurons are inadequate. The initial effort to differentiate human pluripotent stem cells (hPSCs) in the absence of added morphogens generates MAP2+ cortical neurons at day 72. 14 The use of dual SMAD inhibition to drive the differentiation of hPSCs almost completely to the neural lineage represents a significant milestone in the directed differentiation of hPSCs to different types of neurons 15 and neural crest populations. 16 It has been confirmed that dual SMAD inhibition in the absence of other morphogens largely generates neural crest cells. 17 Indeed, adding the WNT inhibitor XAV939 18 to rostralize cell fate 19 and SHH for ventralization 20 generates NKX2.1+ ventral forebrain neuroprogenitors. When these progenitors at day 32 are co-cultured with E13.5 mouse embryonic cortical neuronal cultures or transplanted in mouse cortical slices, they produce cortical GABAergic or cholinergic interneurons. 21 Similarly, the addition of XAV939, the receptor tyrosine kinase inhibitor SU5402, and the MEK inhibitor PD0325901 generates human cortical neurons in the presence of mouse astrocytes. 17 The inclusion of retinoid acid, which is known to caudalize neuroectoderm to the hindbrain fate, 22 generates cortical neurons, 23 albeit with variability caused by uncontrolled dorsal-ventral and rostral-caudal specification that can be suppressed by exogenous activation of WNT signaling. 24
The difficulties in the differentiation of human iPSCs to cortical neurons have stimulated the development of Neurogenin 2 (NGN2)-induced neurons (iNs). 25 Overexpression of exogenous NGN2, a neurogenic transcription factor important for the development of cortical glutamatergic neurons, 26 forces the differentiation of hPSCs to glutamatergic neurons within two weeks. Maturation of the NGN2-iNs to form functional synapses requires co-culture with mouse astrocytes. 25 The ease of the method in producing human neurons has stimulated extensive development using NGN2 in combination with various transcription factors and chemicals to drive the differentiation of hPSCs to many other types of neurons. 27 However, a single-cell RNA sequencing study shows that NGN2-iNs exhibit a gene expression profile very different from that of central neurons and is closer to that of peripheral neurons. 28
The challenges described above stimulated our effort to systematically search for the optimal conditions to specify a dorsal rostral fate on neuroepithelial cells during the differentiation of hiPSCs in dual SMAD inhibitors. Based on our previous success in using developmental biology principles to generate A9 dopaminergic pacemakers, 29 we differentiated hiPSCs in suspension culture to form embryoid bodies (EBs) in the presence of the dual SMAD inhibitors and varying concentrations of the WNT inhibitor XAV939 (XAV) for rostralization and the SHH inhibitor cyclopamine (CP) for dorsalization. After the optimal concentrations of XAV and CP were identified by marker gene expression levels, we searched for their best durations and treatment windows. The systematically optimized method differentiated hiPSCs in 12 days to dorsal forebrain neuroepithelial cells, which generated MAP2+ neurons from day 18 in the presence of the notch inhibitor DAPT and db-cAMP, which promote neuronal differentiation and maturation.17,30 The optimized condition significantly increased the expression of markers for all cortical layers, glutamatergic neurons and GABAergic neurons, while significantly suppressing the expression of forebrain ventral markers at day 30. Further maturation produced mature synapses with punctated juxtaposition of pre- and post-synaptic markers at day 80 and active synaptic transmission at day 95. Most importantly, the hiPSC-derived cortical neurons produced the six main MAPT transcripts from Day 80 at ratios similar to those in postmortem adult human brains.
Methods
Human induced pluripotent stem cells
The normal human iPSC lines used in the study were purchased from California Institute for Regenerative Medicine (CIRM) Human Pluripotent Stem Cell Line Repository [CW70344 (male, 62 years), CW50040 (male, 63 years), CW70305 (female, 56 years), and CW70256 (female, 61 years)] and European Bank for Induced Stem Cells (EBiSC) [STBCi066A (male, 72 years)]. The use of deidentified human cells is not human subject research. The CIRM iPSC lines were generated with non-integrating episomal vectors, while the EBiSC line was generated with non-integrating Sendai viruses. hiPSC cells were cultured on γ-irradiated CF-1 mouse embryonic fibroblast (MEF) in a medium containing DMEM/F12, 20% knockout serum replacement, 0.1 mM β-mercaptoethanol, 1× NEAA, 1× L-glutamine, and 4–8 ng/mL FGF2. The medium was changed daily. Cells were passaged with dispase (1 mg/ml), washed, and replated at a passage ratio of 1:6 weekly. It is imperative to maintain hiPSCs in an undifferentiated state by manually picking morphologically ideal colonies to expand the culture. The quality of hiPSC culture is the most important factor in the differentiation protocol. It is very important to maintain hiPSCs on MEF feeders, as a large-scale study shows that feed-free cultures of hPSCs have more genomic damages and a higher chance of gaining extra chromosome 1q, which enables the abnormal cells to overpopulate the culture only in feeder-free systems. 31 Mycoplasma testing by PCR was conducted on a regular basis to confirm the lack of mycoplasma contamination. 32 Antibiotics were not used in order to expose any problems in cell culture and ensure that aseptic techniques are robust enough for culturing cells up to 150 days. MEF production from mouse embryos is conducted with the approval of University at Buffalo Institutional Animal Care and Use Committee (IACUC).
Differentiation of hiPSCs to dorsal forebrain neuroepithelial cells
hiPSCs were detached from MEF feeders with dispase to generate embryoid bodies (EBs), which were cultured in suspension in a 1:1 mixture of DMEM/F12 and Neurobasal media containing N2 Supplements (1:100), B27 supplements without vitamin A (1:50), ascorbic acid (0.2 mM), 10 μM SB431542 (SB, APExBIO), 5 µM dorsomorphin dihydrochloride (DM, APExBIO), and various concentrations of XAV939 (XAV, APExBIO) and Cyclopamine (CP, APExBIO) for the duration and timing as indicated. It is important to keep the EBs smaller than 50 μm in diameter by breaking larger ones apart through gently pipetting them up and down a few times. Dead cells inside large EBs impair the differentiation. On day 6, EBs were washed with DMEM/F12 once, and then plated on the Matrigel-coated plates evenly for adherent culture. The cells were cultured until day 12, with medium changes every other day.
Differentiation of dorsal forebrain neuroepithelial cells to cortical neurons
On day 12, specified dorsal forebrain neuroepithelial cells were dissociated to single cells with 1 unit/ml Accutase at 37°C for 5 min. Cells were centrifuged at 200× g for 5 min and washed with 10 ml DMEM/F12 twice, and then seeded onto polyornithine/Matrigel-coated plates at a density of 5000–10,000 cells/cm2 in a 1:1 mixture of DMEM/F12 and Neurobasal media that contained N2, B27 without vitamin A (1:50), ascorbic acid (0.2 mM), brain-derived neurotrophic factor (BDNF) (20 ng/ml), glial cell line-derived neurotrophic factor (GDNF) (20 ng/ml), dibutyryl-cAMP (db-cAMP) (0.25 mM) and DAPT (2.5 μM). The ROCK inhibitor Y27632 (20 μM) was added during the first 24 h. At day 16, cells were dissociated and plated again in the same medium. Extra cells not immediately needed were frozen at 1 × 106 cells/ml in STEM-CELLBANKER EX Freezing Medium (amsbio). On day 20, the medium was changed to Neurobasal containing B27 without vitamin A (1:50), BDNF (20 ng/ml), GDNF (20 ng/ml), db-cAMP (0.25 mM) and DAPT (2.5 μM). Half of the medium was changed every other day.
Postmortem human brain tissue
Postmortem brain samples were obtained from NIH NeuroBioBank. Frozen tissue from BA21 region of five normal subjects, 47819 (male, 78 years), 56356 (female, 79 years), 51171 (female, 84 years), 57242 (female, 85 years), and 34913 (male, 85 years) were used for RNA extraction. Frozen tissue from BA21 region of 4 normal subjects, 23983 (male, 79 years), 34913 (male, 85 years), 41250 (female, 86 years), and 47819 (male, 78) were used for western blotting.
Reverse transcription PCR
Total RNA was extracted from cells or tissue using TRIzol (ThermoFisher) and cleaned with a RNeasy kit (QIAGEN). For each sample, 1.0 μg of total RNA was reverse transcribed with the iScriptTM cDNA synthesis kit (BIO-RAD). PCR was performed with Platinum Taq DNA Polymerase (Invitrogen) according to the manufacturer's instructions. The PCR program included an initial denaturation step of 5 min at 94°C, followed by 40 cycles with 60 s at 94°C, 30 s at annealing temperature and 60 s at 72°C, then by extension step of 8 min at 72°C. The annealing temperature for MAPT_012N and MAPT_3R4R primers were 60°C and 65°C respectively. PCR products were separated on 3% agarose gel in TBE buffer at 100 V for 30 min in a cold room. The expected amplicon sizes for MAPT 0N, 1N, 2N, 3R, and 4R were 72 bp, 159 bp, 246 bp, 91 bp, and 184 bp, respectively.
RT-qPCR was performed with IQ SYBR Green Supermix (BIO-RAD) according to the instructions. The PCR program included an initial denaturation step of 4 min at 95°C, followed by 40 cycles of PCR consisting of 30 s at 95°C, 30 s at 60°C and 60 s at 72°C. Average threshold cycle (Ct) values from the triplicate PCR reactions for a gene of interest were normalized against the average Ct values for GAPDH from the same cDNA sample. To calculate the percentage of MAPT exon 10 inclusion, average Ct values of qPCR using interexonic primers MAPT_e9_10 or MAPT_e10_11 were normalized against the average Ct values of qPCR of the same sample using intraexonic primers MAPT_e9. Sequences of PCR primers are listed in Supplemental Table 1.
Immunocytochemistry
Cells cultured on glass coverslips in 12-well or 24-well plates were fixed with 4% paraformaldehyde in PBS for 20 min, permeabilized with 0.1% Triton X-100 in PBS with Ca2+/Mg2+ for 20 min at room temperature, and blocked in 3% BSA in PBS with Ca2+/Mg2+ for 60 min at room temperature. Cells were incubated in the primary antibody in 3% BSA in PBS with Ca2+/Mg2+ at 4°C overnight and washed with PBS with Ca2+/Mg2+ three times, 10 min for each time, followed by the secondary antibody in 4% BSA in PBS with Ca2+/Mg2+ at room temperature for 2 h. Cell nuclei were labeled with 1μg/ml DAPI for 10 min at room temperature. Information on antibodies, chemicals, and growth factors is listed in Supplemental Table 2. Images were collected on a Leica AF6000 inverted fluorescence microscope.
Western blot analysis of tau isoforms
Postmortem brain tissue or iPSC-derived neurons were lysed in buffer A (20 mM Tris-HCl, pH 7.4, 1% Triton X-100, 1 mM PMSF, and a protease inhibitor cocktail) on ice for 30 min. The total lysate was treated with λ-phosphatase (100 units) for 30 min at 30°C, then centrifuged at 12,000 rpm for 10 min at 4°C. The supernatant fraction was collected. The pellet fraction was resuspended in buffer B (20 mM Tris-HCl, pH 7.4, 1% SDS, 1 mM PMSF, and a protease inhibitor cocktail), pipetted up and down 5–6 times, and heated at 95°C for 10 min, cooled to room temperature, and centrifuged at 12,000 rpm for 10 min at room temperature to collect the SDS-soluble fraction. No visible pellet remained. After protein quantification, both the Triton X-100-soluble and Triton X-100-insoluble fractions were separated on 10% SDS-PAGE, and transferred onto 0.4 µm PVDF membranes for western blotting. Tau protein was detected using a 1:2000 dilution of the Tau-1 antibody (MAB3420, Sigma-Aldrich), and a 1:10,000 dilution of HRP-linked anti-mouse IgG secondary antibody (Cell Signaling #7076). The SuperSignal™ West Femto Maximum Sensitivity Substrate was used for visualization. The Tau protein ladder (0.3 μl, rPeptide) contains the six major isoforms of purified recombinant Tau (Tau-352, Tau-383, Tau-381, Tau-410, Tau-412, and Tau-441) produced in E. coli.
Electrophysiological recordings
Whole-cell patch-clamp recordings were performed in iPSC-derived neurons at day 95 or later. Cells were perfused in artificial cerebrospinal fluid (ACSF), consisted of (in mM): 128 NaCl, 1.25 NaH2PO4, 3 KCl, 26 NaHCO3, 1 CaCl2, 4 MgCl4, 10 glucose, 300 mOsm, and 95% O2/5% CO2. Voltage-dependent sodium and potassium currents were recorded with the internal solution containing (in mM): 125 K-gluconate, 10 KCl, 10 HEPES, 0.5 EGTA, 3 Na2ATP, 0.5 Na2GTP and 12 phosphocreatine, pH 7.4, 300 mOsm. Voltage steps from −50 mV to +40 mV lasting 500 ms were used for recordings of voltage-dependent Na+ and K+ currents. For the recording of evoked action potentials, steps of currents from −5 to +65 pA lasting 500 ms were injected into the cell. To record spontaneous action potentials, cells were held in the current-clamp mode with no current injection. An Olympus BX51WI microscope was used to visualize neurons. A MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA) was used to perform patch-clamp recordings. Signals were filtered at 4 kHz and sampled at 100 kHz using a Digidata 1322A analog-digital converter (Axon instruments). Data were processed with pClamp 10.0 (Axon Instruments).
5-ethynyl-2’-deoxyuridine (edu) incorporation
EdU labeling was performed using a kit from Invitrogen. Cells were incubated in media containing EdU (10 µM) for 30 min. After the cells were washed with the same media without EdU three times, they were cultured for another 24 h in the same media without EdU, then fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 for 20 min at room temperature, and stained in the detection cocktail (4 mM copper sulfate, 40 mM sodium ascorbate and 20 μM Alexa Fluor 488 azide, 20 mM Tris, pH 7.6). Cells were counterstained with DAPI to visualize nuclei.
Statistical analysis
GraphPad Prism 6 was used for graph and statistical analysis. All data are expressed as mean ± standard error of measurement. Unpaired student t-test was used for comparing two groups and one-way ANOVA was used for comparing multiple groups more than two.
Results
Optimizing the concentration of rostralizing and dorsalizing morphogens
Our previous success in developing a chemically defined method to differentiate hiPSCs to A9 dopaminergic (DA) pacemakers 29 demonstrates the effectiveness in differentiating hiPSCs through the formation of embryoid bodies (EBs). We used five independent lines of normal hiPSCs, which behaved very similarly in differentiation. The hiPSCs were dissociated from MEF feeders to form EBs in the presence of the dual SMAD inhibitors SB431542 (SB, 10 µM) and dorsomorphin (DM, 5 µM) to neuralize the cells 15 (Figure 1A). Varying concentrations of XAV939 (1.25, 2.5, or 5 μM), a tankyrase inhibitor that blocks WNT signaling, 18 was added at the same time to identify the optimal condition to achieve forebrain specification.19,33 After the EBs were specified for 6 days in suspension culture, they were plated and cultured until day 12 to generate neuroepithelial (NE) cells. RT-qPCR measurement of a representative set of regional markers in human brain development (Figure 1B) showed that dual SMAD inhibition alone increased the expression of the forebrain marker FOXG1 but decreased the dorsal marker EMX1 and increased the ventral marker NKX2.1, while the midbrain marker EN1 and the hindbrain marker HOXA2 were significantly reduced (Figure 1C). This is consistent with a previous study that shows the specification of a forebrain fate by dual SMAD inhibition. 15 The addition of XAV939 (XAV) on top of SB/DM significantly increased forebrain genes (FOXG1, EMX1 and NKX2.1), significantly suppressed EN1, but did not significantly altered the expression of HOXA2 (Figure 1D). Overall, XAV939 at 2.5 μM appeared to be the optimal concentration for inducing the forebrain markers and suppressing EN1.

Identifying the optimal morphogen concentrations for the differentiation of hiPSCs to dorsal forebrain neuroepithelial cells. (A) hiPSCs were cultured in suspension for six days to form embryoid bodies (EBs), which were plated on matrigel and differentiated to dorsal forebrain neuroepithelial (NE) cells in DMEM/F12:Neurobasal (1:1) base medium containing varying concentrations of XAV939 (XAV), Cyclopamine (CP), and the dual SMAD inhibitors SB431542 (SB, 10 μM) and Dorsomorphin (DM, 5 μM) at the indicated time. (B) A schematic of marker gene expression in the human embryonic brain at Carnegie Stage 14. (C) hiPSCs were differentiated in the base medium plus the dual SMAD inhibitors for 6 days and maintained in the base medium to day 12. Expression levels of the indicated genes were measured by RT-qPCR. **p < 0.01, unpaired t-test, versus Day 6, n = 9 from 3 independent experiments with triplicates for each condition. (D,E) hiPSCs were differentiated in the base medium containing the dual SMAD inhibitors and various concentrations of XAV939 (D) or cyclopamine (E) for 6 days and maintained in the base medium to day 12, when expression levels of the indicated genes were measured by RT-qPCR.
In separate experiments, we inhibited Sonic Hedgehog (SHH) signaling with cyclopamine 34 at 1.75, 3.5, or 7 μM to suppress the ventral fate and identify the best condition for dorsalization. 35 We found that cyclopamine (CP) plus SB/DM significantly enhanced the expression of dorsal markers (PAX6, EMX1, TBR1) and significantly suppressed the ventral marker NKX2.1 at 3.5 μM, in comparison to SB/DM only (Figure 1E).
A systematic search for the optimal duration and timing of rostralization and dorsalization
Using XAV939 at the optimal concentration of 2.5 μM, we tested various durations (6, 8, 10, and 12 days) and treatment windows (days 0–10, 2–10, and 4–10) by adding it to the basal condition (SB/DM) (Figure 2A). Treating cells from Days 0–10 induced the highest levels of the forebrain markers FOXG1, EMX1, and NKX2.1, while significantly suppressing the midbrain marker EN1 and not altering the hindbrain marker HOXA2 (Figure 2B). We then optimized the duration (6, 8, and 10 days) and timing (days 0–10, 2–10, and 4–10) of cyclopamine (CP) at the most effective dose (3.5 μM) by adding it to the best condition for XAV939 (D0–10 at 2.5 μM) (Figure 2C). The addition of CP from D0–10 induced the highest levels of the dorsal markers FOXG1, PAX6, and EMX1, significantly increased the dorsal marker TBR1, and suppressed ventral markers NKX2.1 and DLX2 to the lowest level, without significantly affecting the expression of the ventral marker ASCL1 (Figure 2D). Thus, the optimal condition is SB/DM (D0–6) plus XAV (2.5 μM) and CP (3.5 μM) from D0–10.

Differentiation of dorsal forebrain neuroepithelial cells to cortical neurons
Using the optimized condition illustrated in Figure 3A, hiPSCs were differentiated in suspension culture to form EBs. There was no apparent difference in the morphology of EBs at D6 between the basal condition with SB/DM only (Figure 3B) and the optimized condition including XAV and CP (Figure 3B’). After EBs were plated, rosettes generated in the basal condition (Figure 3C) and the optimized condition (Figure 3C’) exhibited a similar morphology with variable sizes at D12. Immunostaining showed that the optimized condition significantly increased the percentage of EMX1+ cells (Figure 3D-D”), did not appreciably affect the very high percentage of PAX6+ cells (Figure 3E-E”), and significantly decreased the percentages of cells expressing NKX2.1 (Figure 3F-F”) or HOXA2 (Figure 3G-G”) (Supplemental Figure 1). The rosettes were picked manually, dissociated, and plated in media containing neurotrophic factors GDNF and BDNF, the notch inhibitor DAPT to promote neuronal differentiation, 30 and db-cAMP to facilitate neuronal maturation.17,30 After two additional passages to maintain cell density suitable for differentiation, neurons were maturated in a neuronal culture medium (Figure 3A).
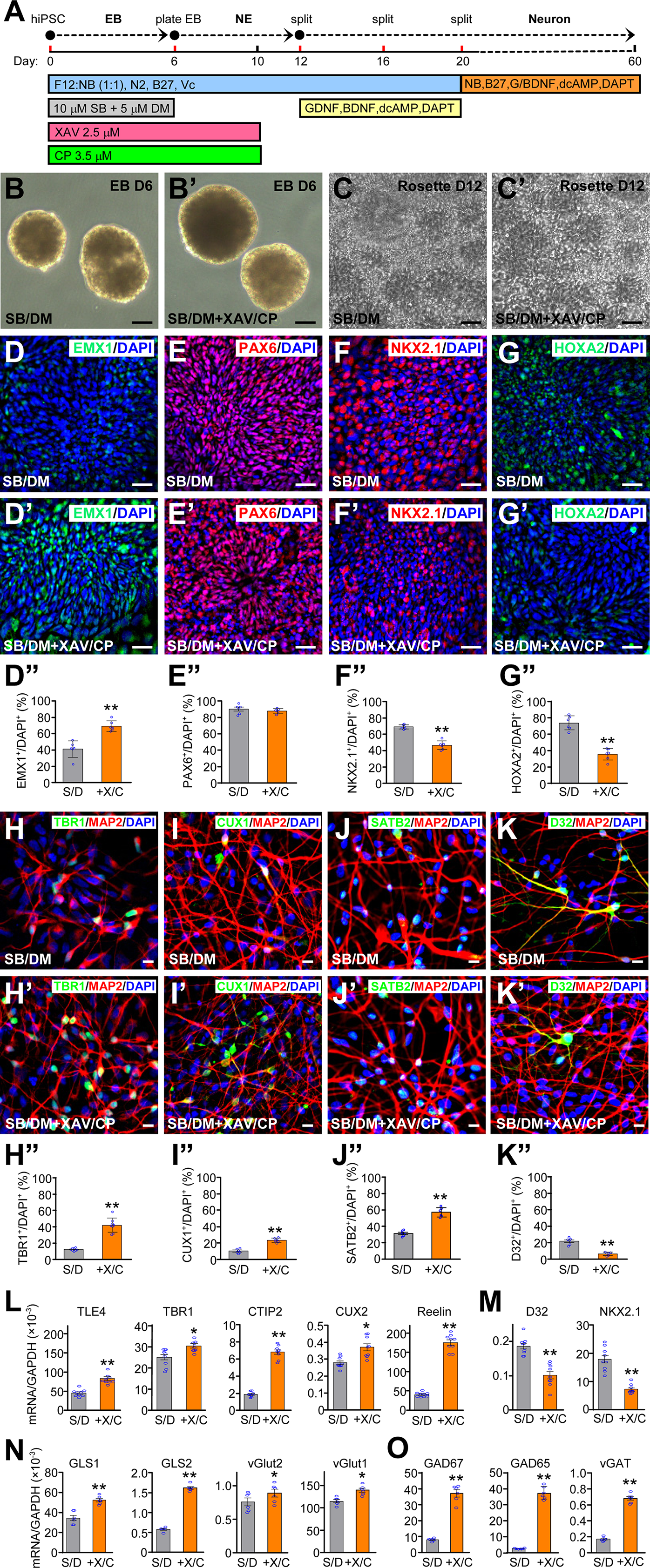
Differentiation of hiPSCs to cortical neurons with the optimized condition. (A) hiPSCs were differentiated through EBs to neuroepithelial (NE) cells then to cortical neurons as indicated. (B, B’) EBs at day 6 in the presence of SB and DM only (B) or plus XAV and CP (B’). Bars, 10 μm. (C, C’) NE cells in rosettes at day 12 in the presence of SB and DM only (C) or plus XAV and CP (C’). Bars, 10 μm. D-G’’ NE cells generated in the presence of SB and DM only (D-G) or plus XAV and CP (D’-G’) were stained at day 12 for EMX1 (D, D’), PAX6 (E, E’), NKX2.1 (F, F’), and HOXA2 (G, G’) and quantified (D”-G”). Bars, 10 μm. **p < 0.01, unpaired t-test, n = 6 from 3 independent experiments, with 2 coverslips for each condition. (H-K”) Cells differentiated in the presence of SB/DM only (H-K) or plus XAV/CP (H’-K’) were co-stained for TBR1 (H, H’, H”) at day 30, CUX1 (I, I’, I”), SATB2 (J, J’, J”), and DARPP32 (D32) (K, K’, K”) at day 70, and quantified (H”-K”). Bars, 10 μm. **p < 0.01, unpaired t-test, n = 6 from 3 independent experiments, with 2 coverslips for each condition. (L-O) Expression levels of the indicated cortical layer markers (L), ventral markers (M), glutamatergic neuronal markers (N) and GABAergic neuronal markers (O) at day 30. *p < 0.05, **p < 0.01, unpaired t-test, n = 9 from 3 independent experiments with triplicates for each condition.
The optimized condition produced a significantly higher percentage of cells expressing the cortical deep layer marker TBR1 (Figure 3H-H”), the intermediate layer marker CUX1 (Figure 3I-I”), and the upper layer marker SATB2 (Figure 3J-J”), while significantly decreasing the percentage of cells expressing the ventral neuronal marker DARPP32 (Figure 3K-K”) (Supplemental Figure 1). We confirmed the immunostaining results by RT-qPCR measurements of various cortical layer markers at day 30. The expression levels of TLE4 (layer VI), TBR1 (layer V-VI), CTIP2 (layer V), CUX2 (layer II-IV), Reelin (layer I) were significantly increased in the optimized condition (Figure 3L), while ventral markers, such as DARPP32 and NKX2.1, were significantly suppressed (Figure 3M). Furthermore, glutamatergic neuronal markers, such as GLS1, GLS2, vGlut1, and vGlut2 (Figure 3N), and GABAergic neuronal markers, such as GAD67, GAD65, and vGAT (Figure 3O) were significantly increased in the optimized condition.
Formation of cell aggregates of neurons and radial glia-like cells
In the differentiation of dorsal forebrain neuroepithelial cells from day 12 onwards, cells extended processes with increasing length and complexity over time. At day 18, neurons with some neurites were evenly dispersed in culture (Figure 4A). Starting around day 25, some cells began to form aggregates (Figure 4B), which increased in size at day 33 (Figure 4C) and beyond. Immunostaining of cells from days 18 to 33 showed increased production of MAP2+ neurons, which surrounded increasingly more Ki67+ proliferating cells in the aggregates (Figure 4A’-C’) (Supplemental Figure 2A’-C’). Some cells in the aggregates at day 33 expressed the neural stem cell marker SOX1 and the radial glial marker vimentin (Vim) (Figure 4D-E) (Supplemental Figure 2D-E). EdU labeling at day 35 showed that most of the proliferating cells were PAX6+ (Figure 4F-F’) and SOX2+ (Figure 4G-G’). There appeared to be more of these proliferating cells in the aggregates (Figure 4F’ and G’) than among the non-aggregated cells (Figure 4F and G) (Supplemental Figure 2F-G’).

Properties of cell aggregates formed in hiPSC-derived cortical neuronal cultures. (A-C’) Phase contrast images (A-C) and immunostaining (A’-C’) of iPSC-derived cortical neuronal cultures at the indicated days showed the formation of cell aggregates containing MAP2+ neurons and ki67+ mitotic cells. (D, E) Aggregates at day 33 contained cells expressing the neural stem cell marker SOX1 (D) and the radial glial marker vimentin (E). F-G’ In cultures incubated with EdU for 24 h at day 35, both dispersed areas (F, G) and aggregates (F’, G’) contained PAX6+ (F, F’) or SOX2+ (G, G’) cells labeled with EdU. H-M’ Aggregates at day 33 were picked, dissociated, cultured in a new well to day 45 (H). After removing the aggregates at day 33, the remainder of the culture was imaged at day 45 (H’). Neurons derived from dissociated aggregates (I-M) or the remainder neurons (I’-M’) were stained as indicated for various cortical layer markers at day 45 or neurotransmitter markers at day 80. (N) Expression levels of the indicated marker genes in both preparations at day 45.
The presence of proliferating cells expressing markers of radial glial cells and neural stem cells prompted us to pick and dissociate these aggregates with Accutase at day 33 for culture on Matrigel-coated plates. The dissociated cells became neurons within 7 days and extended increasingly complex processes. At day 45, neurons derived from dissociated aggregates (Figure 4H) had morphology very similar to that of remainder neurons after the aggregates were picked off (Figure 4H’). Both preparations exhibited very similar expression of cortical layer markers TBR1, CUX1, and SATB2 in MAP2+ neurons at day 45 (Figure 4I-K’) (Supplemental Figure 3) and the glutamatergic (vGlut2) or GABAergic (GABA) neuronal marker in MAP2+ neurons at day 80 (Figure 4L-M’) (Supplemental Figure 4). RT-qPCR experiments confirmed that the two preparations at day 45 had the similar expression levels of various cortical layer markers (TLE4, CTIP2, CUX2, and Reelin), glutamatergic neuronal markers (GLS1, GLS2, vGlut1, and vGlut2), GABAergic neuronal markers (GAD65, GAD67, and vGAT), and mature neuronal markers (MAP2 and NeuN) (Figure 4N).
Synapse formation and synaptic transmission in iPSC-derived cortical neurons
In mature iPSC-derived cortical neurons at day 80, we observed punctated localization of vesicular glutamate transporter 1 (vGlut1) and vGlut2 in glutamatergic neurons (Figure 5A, B). GABAergic neurons expressing GABA or GAD67 were also found in the culture (Figure 5C, D). Punctated localization of the post-synaptic markers GluR1 and PSD95, as well as the pre-synaptic markers SNAP25 and Syntaxin 1A (STX1A), were found in juxtaposition (Figure 5E, F) (Supplemental Figure 5), suggesting the formation of synapses. The culture also contained GFAP+ astrocytes (Figure 5G) (Supplemental Figure 5). Cell counting showed that 28.1 ± 3.5% of MAP2+ neurons were vGLUT2+ glutamatergic neurons, while 25.4 ± 3.2% MAP2+ neurons were GAD67+ GABAergic neurons (Figure 5H). Among all DAPI+ cells, 59.1 ± 3.0% was MAP2+ neurons, and 6.3 ± 0.9% was GFAP+ astrocytes (Figure 5I). Electrophysiological recordings of these neurons at day 95 showed voltage-gated Na+ currents and voltage-gated K+ currents (Figure 5J), evoked action potentials in response to current injections (Figure 5K), spontaneous excitatory postsynaptic currents (Figure 5L), and spontaneous action potentials (Figure 5M). These results show that the iPSC-derived cortical neurons have active synaptic transmission.
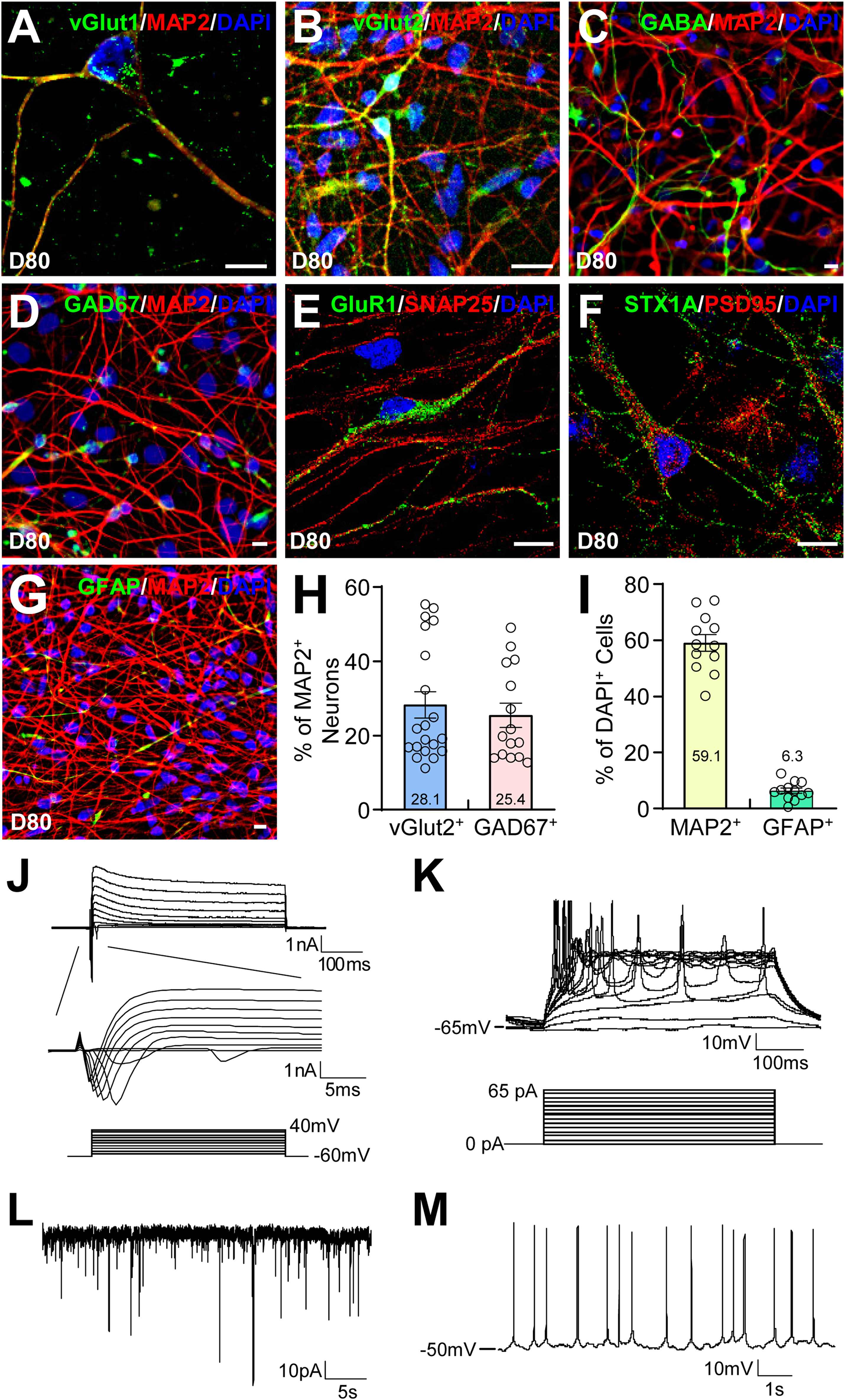
Functional characterization of hiPSC-derived cortical neurons. (A-G) hiPSC-derived cortical neuronal cultures were stained as indicated at day 80. Bars, 10 μm. (H) The percentage of vGLUT2+ glutamatergic neurons or GAD67+ GABAergic neurons in MAP2+ neurons was quantified by cell counting. (I) The percentage of MAP2+ neurons or GFAP+ astrocytes in DAPI+ cells was quantified by cell counting. (J-M) Electrophysiological recordings of the neurons at day 95 showed that they had voltage-gated Na+ current and K+ currents (J), evoked action potentials (K), spontaneous excitatory postsynaptic currents (L), and spontaneous action potentials (M).
Expression of the six main MAPT transcripts in a developmentally regulated manner
We designed PCR primers to examine the expression of 0N, 1N, 2N, as well as 3R and 4R AS isoforms of Tau (Figure 6A). Total RNA isolated from cortical neurons differentiated for various durations from three independent lines of hiPSCs and frozen postmortem cortical tissue from BA21 area of five normal adults were analyzed by RT-PCR. Both 1N and 2N transcripts were expressed from Day 80 and became much more prominent at Days 100 and 120 in one line of cortical neurons. The expression levels were variable between different lines of cortical neurons, analogous to the individual variations in postmortem cortical tissue (Figure 6B). The 4R transcript was expressed at various levels in the three lines of cortical neurons, as early as Day 50 in two lines. At Day 80, the 4R Tau transcript was expressed at approximately equal ratio to the 3R transcript, very similar to the situation in adult postmortem brain tissue (Figure 6C). To confirm the expression of 4R Tau, we performed RT-qPCR to quantify the ratio of 4R versus 3R Tau transcripts using interexonic primers spanning exons 9 and 10 and intraexonic primers within exon 9 (as an internal control) (Figure 6A). Indeed, the 4R/3R ratios in the same samples (Figure 6D) were consistent with quantification of the RT-PCR gel (Figure 6C). Another pair of interexonic primers spanning exons 10 and 11 (Figure 6A) were used to confirm the results further (Figure 6E). Immunostaining of cortical neurons with the 4R-specific ET3 antibody 13 showed prominent expression of 4R Tau proteins at Day 149, but not at Day 60 (Figure 6F). We lysed four lines of iPSC-derived cortical neurons (at D95 to D110) along with postmortem cortical tissue from four normal adults (ages 78 to 86 years) in a buffer containing 1% Triton X-100. After the total cell lysate was treated with λ-phosphatase to dephosphorylate Tau, the supernatant fraction was collected by centrifugation. The pellet fraction was dissolved in a buffer containing 1% SDS. Western blotting with the Tau-1 antibody showed the presence of the six isoforms of Tau, most prominently in the Triton X-100-insoluble, but SDS-soluble, fraction (Figure 6G). In the Triton X-100-insoluble fraction, there appeared to be an approximately equal ratio of 4R and 3R Tau isoforms, with the most abundant being 1N4R and 1N3R, as judged by comparing the bands with the recombinant Tau ladder (Figure 6G).
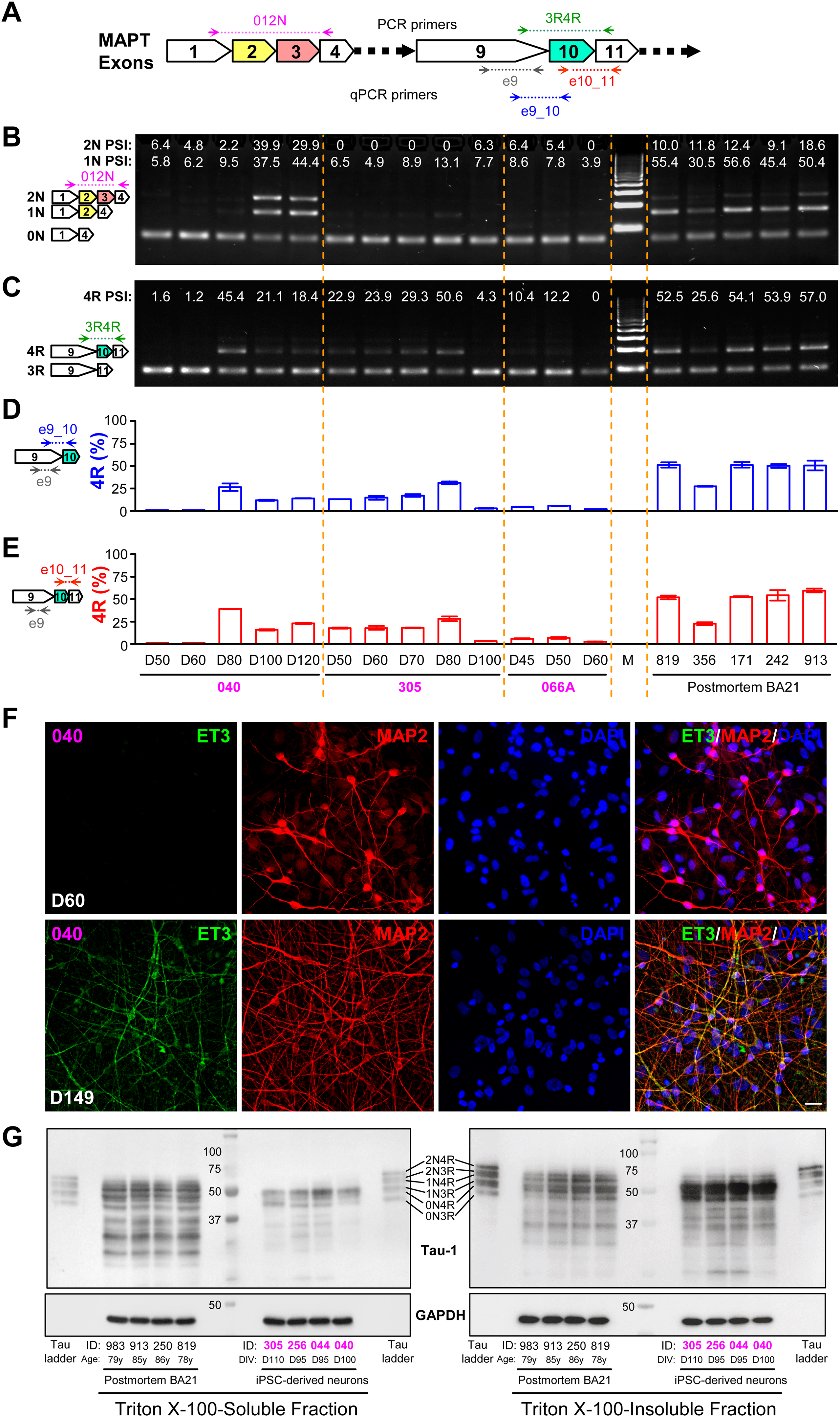
Expression of the six Tau isoforms in hiPSC-derived cortical neurons. (A) Schematic representation of the three major alternative exons (2, 3, and 10) expressed in the adult human brain and their neighboring constitutive exons of the MAPT gene. Locations of PCR and qPCR primers for the detection of 0N, 1N, 2N transcripts, as well as 3R and 4R transcripts are shown. (B, C) RT-PCR detection and quantification of MAPT 0N, 1N, 2N transcripts (B), as well as 3R and 4R transcripts (C) in total RNA isolated from the indicated hiPSC-derived cortical neurons at different days and postmortem adult human brain tissue (BA21 area). (D, E) Using intraexonic primers e9 as an internal control, the percentage of 4R transcripts was quantified in the same set of samples using interexonic primers e9_10 (D) or e10_11 (E). (F) hiPSC-derived cortical neurons at D60 or D149 were stained for the 4R-specific antibody ET3, as well as MAP2 and DAPI. (G) Triton X-100–soluble and –insoluble fractions of total lysates from the indicated materials were separated on SDS-PAGE along with the six recombinant Tau isoforms (Tau ladder) and blotted with Tau-1 antibody. Subject IDs in the Methods section were abbreviated to last three digits to save space, with postmortem tissues in black font and iPSC-derived neurons in magenta.
Discussion
In this study, we directed the differentiation of hiPSCs to cortical neurons by systematically searching for the optimal concentration, duration, and timing of morphogens for the rostral-caudal and dorsal-ventral specification of EBs being neuralized by dual SMAD inhibition. We found that inhibiting WNT signaling using XAV939 18 at 2.5 μM from days 0–10 in conjunction with blocking SHH signaling by cyclopamine 34 at 3.5 μM from days 0–10 in the presence of the SMAD inhibitors SB431542 (10 µM) and dorsomorphin (5 µM) differentiated hiPSCs to dorsal forebrain neuroepithelial cells. We used a panel of forebrain (FOXG1, EMX1, NKX2.1), midbrain (EN1), and hindbrain (HOXA2) markers to search for the optimal condition for forebrain specification and a panel of dorsal (PAX6, EMX1, TBR1) or ventral (NKX2.1, ASCL1, DLX2) markers to optimize dorsalization. The properly specified dorsal forebrain neuroepithelial cells were differentiated to cortical neurons in the presence of the NOTCH inhibitor DAPT to promote neuronal differentiation, db-cAMP to facilitate neuronal maturation, and neurotrophic factors (BDNF and GDNF) to enhance neuronal survival and maturation.17,30
This method generates MAP2+ neurons at day 18 (Figure 4A’) without the need for FACS sorting or co-culture with mouse astrocytes or mouse cortical neurons. Previous studies have reported the generation of MAP2+ neurons at day 30 after FACS enrichment of CD44−/CD184+/CD24+ neurons, 21 or at day 35 in the presence of mouse astrocytes. 17 Without using mouse astrocytes, differentiation of hPSCs in dual SMAD inhibitors and retinoid acid generates Tuj1+ cortical neurons after day 20, while MAP2+ mature neurons are produced later at an unspecified date.23,24 Since it takes 72 days to generate MAP2+ neurons in spontaneous differentiation without dual SMAD inhibition, 14 it seems plausible that the speed of generating MAP2+ neurons may reflect how well neuronal fate is specified during hPSC differentiation. Improved understanding of human fetal brain development through single-cell RNAseq, multiplexed FISH, single-cell Stereo-seq, and other approaches36–39 will provide valuable information on additional marker genes and morphogens that can be used to refine the differentiation protocol in future studies.
The presence of GABAergic neurons in our cortical neuronal culture (Figure 5C, D) suggests that the dorsal-ventral specification is an operational compromise that does not exclude the production of ventral forebrain progenitors, which generate GABAergic neurons that migrate tangentially to the cortex in vivo. 40 We found that inhibiting SHH signaling by cyclopamine (CP) at 3.5 μM significantly suppressed the ventral marker NKX2.1. Further increase of CP did not decrease NKX2.1 expression (Figure 1E). The ideal duration and timing of CP treatment was days 0–10, which significantly suppressed ventral markers NKX2.1 and DLX2, but not ASCL1 (Figure 2D). As these ventral markers were not completely suppressed, the production of GABAergic neurons is expected and operationally makes the mixed neuronal populations more useful for functional studies. It would be highly challenging to generate in vitro the right mixture of hiPSC-derived cortical glutamatergic neurons and GABAergic neurons that can recapitulate features associated with the radial migration of glutamatergic neurons from the subventricular zone and the tangential migration of GABAergic neurons from the ventral forebrain. The development of various brain organoids has an intractable problem that cells inside the organoids are dead as the organoids grow to several millimeters in size over the months required to generate neurons from hPSCs. 41
The generation of cell aggregates containing both MAP2+ neurons and mitotic cells expressing markers for radial glial cells and neural stem cells (Figure 4) is an interesting feature of this protocol. Dissociated cells from the aggregates at day 33 became neurons within 7 days and exhibited complex neuronal morphology indistinguishable from the original undissociated neurons at day 45. Importantly, both the dissociated neurons and the undissociated neurons showed very similar expression of markers for all cortical layers, glutamatergic and GABAergic neurons, and mature neurons. Future studies are needed to understand the mechanistic details on how these mitotic cells rapidly differentiate into cortical neurons of the same properties as the undissociated neurons. Nevertheless, the ability to generate a large quantity of cortical neurons from these aggregates is a benefit to many applications.
The presence of the six major AS transcripts of MAPT in our iPSC-derived cortical neurons sets this differentiation method apart from all previous studies, which have failed to generate 4R Tau even after one year of differentiation and have not examined whether 1N and 2N Tau can be produced.8–11 It appears that by following developmental biology principles, our differentiation protocol properly models the generation and maturation of cortical neurons in the human brain. The appearance of equal ratio of 4R/3R transcripts from Day 80 and the presence of 1N and 2N transcripts from Day 100 suggest that the development of our iPSC-derived cortical neurons is significantly accelerated, as these transcripts are found in the adult, but not fetal, human brain. 7 Western blotting confirmed the presence of the six Tau protein isoforms around Day 100, most prominently in the Triton X-100-insoluable fraction. It suggests the formation of Tau aggregates. The ratio of 4R and 3R Tau proteins appeared to be equal, corroborating PCR analyses of MAPT transcripts. Thus, these iPSC-derived cortical neurons effectively model the production of Tau isoforms to a pattern quite similar to that in postmortem brain tissue from adults aged 78 to 86 years. Thus, the method would be very useful in modeling various tauopathies with alterations in the 4R/3R ratio in future studies.
Since almost all differentiation protocols use DAPT, db-cAMP, BDNF and GDNF at similar concentrations to differentiate neuroepithelial cells to neurons and to promote neuronal maturation, it is likely that the way we specified EBs and neuroepithelial cells is the key to our success in producing cortical neurons with the six main isoforms of Tau. Although much work is needed to identify key contributors to the success, our method will greatly facilitate the in vitro study of human cortical neurons under the normal condition and in brain disorders ranging from Alzheimer's disease to autism.
Supplemental Material
sj-docx-2-alz-10.1177_13872877251334831 - Supplemental material for Generation of human induced pluripotent stem cell-derived cortical neurons expressing the six tau isoforms
Supplemental material, sj-docx-2-alz-10.1177_13872877251334831 for Generation of human induced pluripotent stem cell-derived cortical neurons expressing the six tau isoforms by Houbo Jiang, Zichun Xiao, Komal Saleem, Ping Zhong, Li Li, Gaurav Chhetri, Pei Li, Zhongjiao Jiang, Zhen Yan and Jian Feng in Journal of Alzheimer's Disease
Supplemental Material
sj-pdf-3-alz-10.1177_13872877251334831 - Supplemental material for Generation of human induced pluripotent stem cell-derived cortical neurons expressing the six tau isoforms
Supplemental material, sj-pdf-3-alz-10.1177_13872877251334831 for Generation of human induced pluripotent stem cell-derived cortical neurons expressing the six tau isoforms by Houbo Jiang, Zichun Xiao, Komal Saleem, Ping Zhong, Li Li, Gaurav Chhetri, Pei Li, Zhongjiao Jiang, Zhen Yan and Jian Feng in Journal of Alzheimer's Disease
Footnotes
Acknowledgments
We thank Emily Fisher for reading the manuscript. Human postmortem brain tissues are obtained from the NIH NeuroBioBank. Human iPSCs are obtained from the California Institute for Regenerative Medicine (CIRM).
ORCID iDs
Ethical considerations
The use of deidentified human cell lines and human postmortem materials is not human subject research under National Institute of Health guidelines. The use of mouse embryos to generate mouse embryonic fibroblasts is conducted with the approval of University at Buffalo Institutional Animal Care and Use Committee.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contributions
Houbo Jiang (Conceptualization; Data curation; Formal analysis; Investigation; Writing – original draft; Writing – review & editing); Zichun Xiao (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing – review & editing); Komal Saleem (Data curation; Formal analysis; Investigation; Methodology; Validation; Writing – original draft; Writing – review & editing); Ping Zhong (Data curation; Formal analysis; Investigation; Writing – review & editing); Li Li (Data curation; Formal analysis; Investigation; Methodology; Writing – review & editing); Gaurav Chhetri (Data curation; Formal analysis; Investigation; Methodology; Writing – review & editing); Pei Li (Data curation; Formal analysis); Zhongjiao Jiang (Data curation; Writing – review & editing); Zhen Yan (Funding acquisition; Methodology; Project administration; Resources; Software; Supervision; Validation; Writing – review & editing); Jian Feng (Conceptualization; Funding acquisition; Project administration; Resources; Supervision; Writing – original draft; Writing – review & editing).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work is supported by the National Institutes of Health grants NS113763 (JF), AG079797 (JF and ZY), NS127728 (JF and ZY), and AG067597 (JF and ZY).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available on request from the corresponding author.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.