
Abstract
Recent findings suggest that neurodevelopment plays a critical role in Huntington's Disease (HD) pathogenesis. This review integrates data from human studies of children and young adults at risk for HD (the Kids-HD study) with the theory of antagonistic pleiotropy (AP), which posits that genes promoting early-life advantages may confer late-life risks. Longitudinal imaging of gene-expanded (GE) children and adolescents shows that mHTT is associated with larger cortical volumes, enhanced surface morphology, and superior cognitive performance—decades before clinical onset. However, this early benefit is paired with accelerated striatal decline, suggesting that mHTT drives an early “ability” that transitions into a “liability.” Vertex-wise analyses reveal cortical enlargement in regions with dense glutamatergic projections to the striatum, implicating excitotoxicity as a mechanism linking development to degeneration. This pleiotropic pattern parallels evolutionary models, where genes like HTT may have an evolutionary trade-off where genes supporting growth and reproduction are favored over those that serve long-term somatic maintenance, leaving cells with diminished repair capacity and resulting in an accelerated aging process. Altogether, these findings support a novel framework in which mHTT accelerates both brain maturation and neurodegeneration, offering new insights into HD biology and therapeutic targets.
Plain language summary
Although Huntington's Disease (HD) is conceptualized as a progressive degenerative brain disease, many studies have shown that the beginnings of the disease may occur during brain development. The gene that causes HD is known to be important in helping to shape brain development. This article reviews findings from human studies of children at risk for HD and discusses how the gene may be driving the development of a superior brain early in life that is built in such a way in which it is then vulnerable to accelerated aging later in life. This is termed “antagonistic pleiotropy” in which a gene can create both advantage and disadvantage. This process is also discussed in the context of evolutionary biology where antagonistic pleiotropy is a leading theory of both human brain evolution as well as aging.
A growing body of research supports the view that neurodevelopment plays a critical role in the pathoetiology of Huntington's disease (HD), the central theme of this special issue. This article reviews key aspects of HD neurodevelopment, emphasizing insights from human studies and the framework of antagonistic pleiotropy (AP). AP is a foundational theory in aging and evolutionary biology, suggesting that genes conferring early-life advantages may also promote late-life vulnerabilities. In the context of HD, it has been hypothesized that the mutant huntingtin (mHTT) gene contributes to enhanced neurodevelopment early in life, but at the cost of increased susceptibility to neurodegeneration later, a concept akin to accelerated aging (Figure 1).

Higher CAG repeats within HTT may confer neurodevelopmental advantages early in life that predispose subjects with HD to neurodegeneration later in life, in line with the theory of antagonistic pleiotropy.
Advantageous or abnormal development?
Wild-type huntingtin (HTT) is essential for numerous neurodevelopmental processes.1,2 In HD, research has largely focused on how mHTT alters brain development. A robust body of literature, spanning molecular, cellular, and animal models, demonstrates that mHTT can lead to developmental abnormalities. Many of these findings are reviewed in detail in this issue.
The prevailing view is that mHTT causes subtle developmental disruptions that are initially compensated for but ultimately render the brain more vulnerable to degeneration. 2 However, emerging evidence challenges this deficit-based model, suggesting that mHTT may instead promote certain advantageous neurodevelopmental features. This distinction between abnormal versus advantageous development is nuanced, as many findings reflect differences whose long-term significance remains unclear. Whether these developmental changes are ultimately detrimental or adaptive is still an open question.
The power of human studies
Despite the wealth of high-quality data generated from basic science models, studying disease processes directly in humans remains the most powerful approach—particularly for disorders of the human brain. While many physiological systems are well-modeled in animals, the human brain has undergone substantial evolutionary changes that distinguish us from our closest evolutionary relatives, the great apes. It is not the physiology of organs like the kidney or liver that sets humans apart, but the exceptional complexity of our brains. Consequently, some neurological and psychiatric diseases may only be fully understood through direct human research.
The HD field has capitalized on this opportunity through a series of influential neuroimaging studies. Beginning in 2001, the PREDICT-HD study enrolled presymptomatic (preHD) adults, 3 followed by large-scale efforts such as TRACK-HD and IMAGE-HD.4,5 These studies demonstrated that structural and functional brain changes in gene-expanded individuals can precede motor onset by as much as 20 years. Coupled with evidence linking these changes to biomarkers of neuronal injury, such as elevated neurofilament light (NfL), these findings established a prolonged preclinical phase of neurodegeneration. 6
However, these discoveries raised new questions: what precedes the degenerative phase? The Young Adult Study (YAS) examined individuals at an average of 23.6 years (SD = 5.8) from predicted motor onset. It found, smaller putamen volumes in preHD subjects, but mostly similar brain volumes compared to controls. Also, like previous studies of preHD individuals, they found evidence of elevated NfL, 7 supporting the notion that degeneration begins around two decades before onset. Yet, this also underscored the limitation: to investigate potential developmental origins of HD, we must study individuals even further from onset, namely, children and adolescents at risk.
The Kids-HD study was launched at the University of Iowa in 2009, enrolling at-risk youth aged 6–25 years. 8 In 2019, it was expanded into the multi-site ChANGE-HD study (Child to Adult Neurodevelopment in Gene-Expanded HD), running through 2026. Participants are genotyped and classified as gene-expanded (GE, CAG > 36) or gene non-expanded (GNE, CAG < 35). The average estimated years to onset in this cohort is approximately 35 years, with some participants more than 60 years from predicted onset, offering an unprecedented window into the earliest phases of the HD disease trajectory.
Initial analyses focused on GNE individuals to examine how CAG repeat length influences brain development. Consistent with the established role of wild-type HTT in neurodevelopment, it was hypothesized that increasing CAG repeats within the non-pathogenic range might confer beneficial effects. Others have reported increasing CAG repeats below disease threshold resulting in increasing volumes of the pallidum. 9 In the KidsHD study, analyses revealed that higher CAG repeats (below disease threshold) were associated with larger cortical volumes and higher IQ scores. 10 Subsequent investigations extended this analysis across the full CAG repeat spectrum, assessing not only how repeat length affects brain structure and function, but also how it influences developmental trajectories over time.
Modeling age trajectories
The Kids-HD study utilized an accelerated longitudinal design (ALD), the gold standard for evaluating brain development in children, which combines cross-sectional and longitudinal data to model age-related changes across a broad developmental window. 11
Figure 2 illustrates modeled age trajectories across four cognitive domains (language, memory, executive function, and visual perception) with curves stratified by CAG repeat length. These analyses revealed a striking pattern: individuals with CAG repeats between 37 and ∼43 exhibited above-average cognitive performance compared to controls. 12 In contrast, individuals with longer CAG repeats (≥45) showed trajectories that were declining in function after age 15 likely representing changes due to the degenerative phase of the disease.
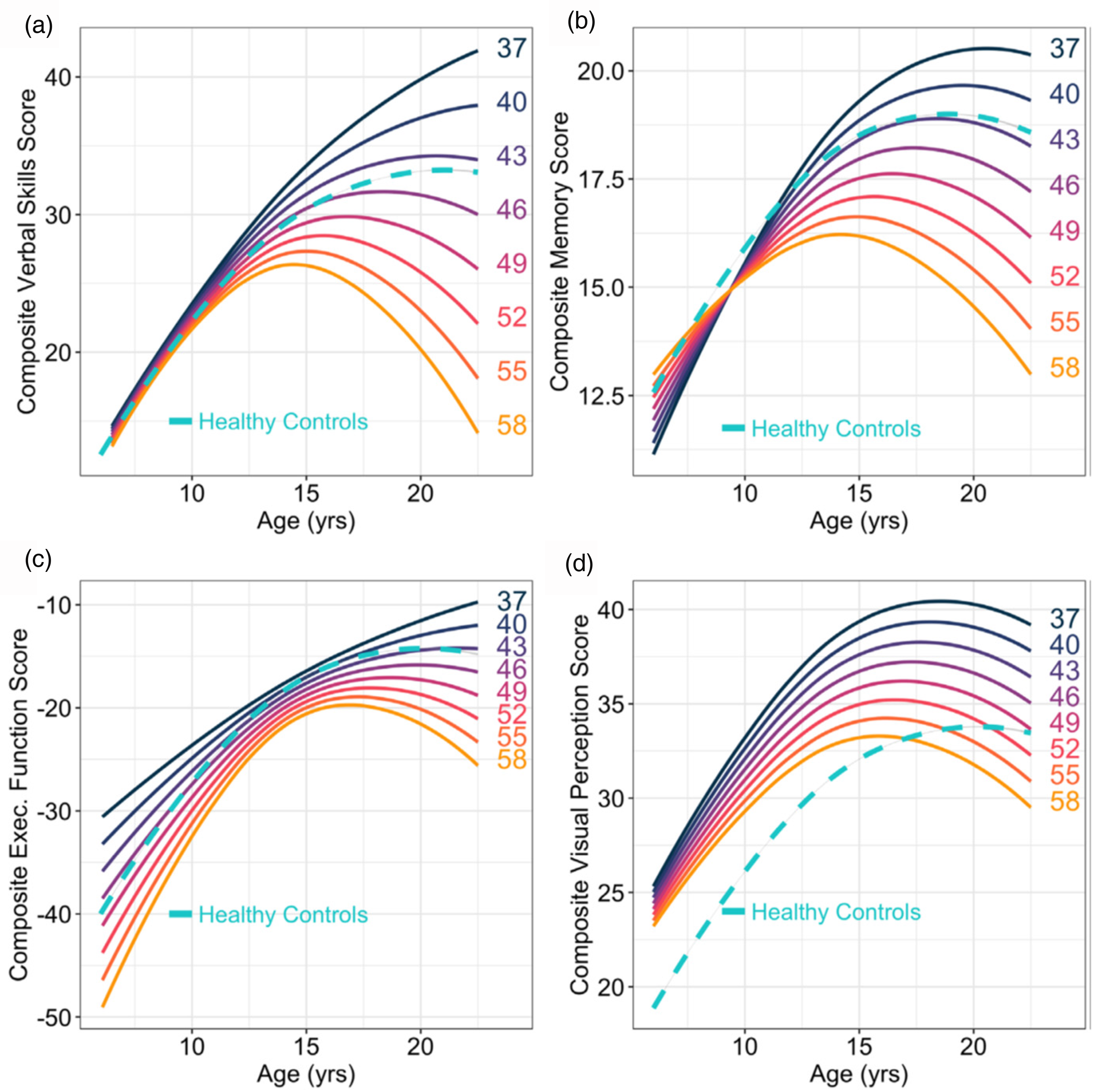
Predicted developmental trajectories of (a) language development, (b) memory, (c) executive function, and (d) visual perception (y-axes) across age (x-axes) separated by CAG repeat length (solid, colored lines). The dashed, turquoise lines represent the developmental trajectories of GNE controls.
These findings underscore that HTT profoundly influences neurodevelopment, and critically, that its effects are CAG repeat-length dependent. This mirrors the well-established relationship between CAG repeat length and age of motor onset, which enables calculation of Predicted Age of Onset (PAO). 13 By subtracting a subject's current age from their PAO, predicted Years to Onset (YTO) can be derived.
Given that adult preHD studies indicate the neurodegenerative process begins up to 20 years before motor onset, it is crucial to understand what occurs even earlier during the developmental phase. However, the strong impact of CAG repeat length on developmental trajectories complicates modeling with age alone. Figure 3 illustrates this point by evaluating HD stages for 3 individuals with varying CAG repeats. A 15 year-old with CAG 42 may still be in the pre-degenerative, developmental phase (Stage 0), whereas a peer with CAG 55 may already be well into the early degenerative period, just 11 years from their predicted motor onset. Thus, simple age-based models fail to account for the heterogeneity introduced by repeat length. The solution is to model developmental change using YTO, which integrates both age and genetic burden, offering a more precise framework for capturing early, HD-related brain changes.
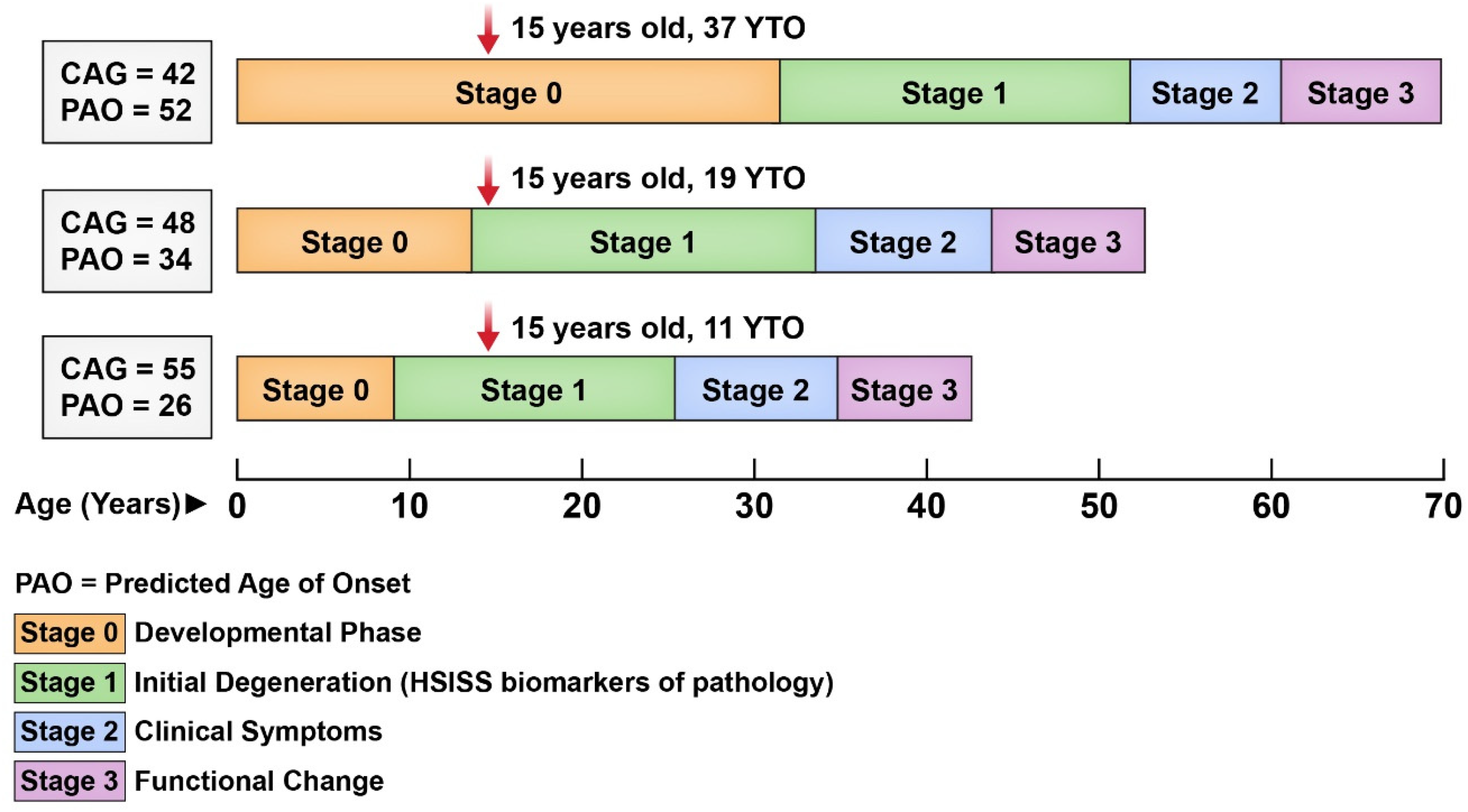
Illustration of HD stages across the CAG repeat spectrum.
Modeling years to onset (YTO)
Our most recent analysis of the Kids-HD dataset employed a YTO model to assess changes across decades of the disease process with 136 GE observations compared to 191 GNE observations, all aged 6–21 years. 14 Measures included brain structure (MRI-derived regional volumes and cortical morphology) and brain function (cognitive, motor, and behavioral performance). Figures 4–6 display trajectories across a wide YTO range—from >50 years before predicted motor onset to <10 years from onset.

YTO-dependent brain functional changes. Predicted years-to-onset (YTO) dependent trajectories of (a) General Ability Index, (b) Behavior Composite, and (c) PANESS Score(motor function) in the gene-expanded (GE) participants throughout the premanifest course of Huntington disease. In each plot, the solid-colored line depicts the best predicted fit with shaded region illustrating the 95% confidence interval. On the Y-axis, the demographically adjusted values represent the difference between unadjusted functional scores in GE and the age/sex-based predicted functional scores from the gene-non-expanded (GNE) model. The horizontal black dashed line across YTO (x-axis) represents the average GNE control participants scores.

YTO-dependent brain structural changes predicted years-to-onset (YTO) dependent trajectories of (a) whole brain, (b) cerebral, (c) cortical gray matter, (d) cerebral white matter, (e) cerebellum, (f) Striatum, (g) globus Pallidus, and (h) thalamus in the gene-expanded (GE) participants throughout the premanifest course of Huntington disease. In each plot, the solid-colored line depicts the best predicted fit with shaded region illustrating the 95% confidence interval. On the Y-axis, the demographically adjusted volumes (ml) represent the difference between unadjusted volumes in GE and the age/sex-based predicted volumes from the gene-non-expanded (GNE) model. The horizontal black dashed line across YTO (x-axis) represents the GNE control participants.
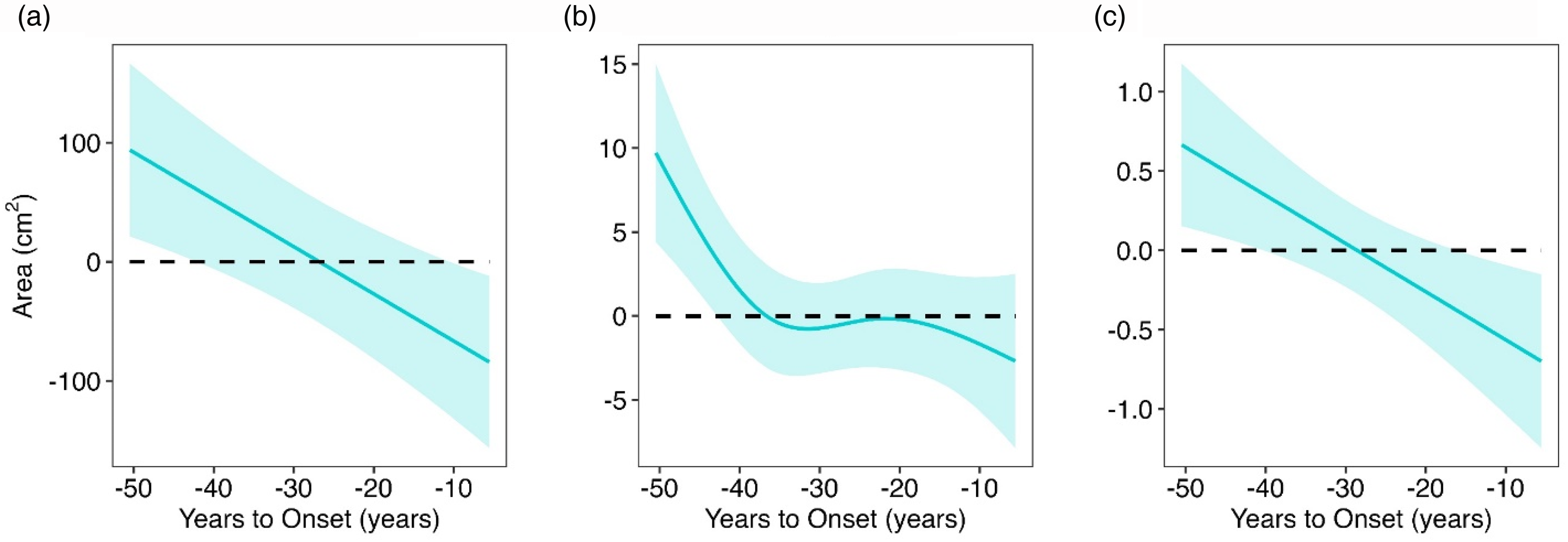
YTO-dependent cortical morphometric changes/ predicted years-to-onset (YTO) dependent trajectories of (a) cortical surface area, (b) cortical folding Index, and (c) cortical curvature Index in the gene-expanded (GE) participants throughout the premanifest course of Huntington disease. In each plot, the solid-colored line depicts the best predicted fit with shaded region illustrating the 95% confidence interval. On the Y-axis, the adjusted values represent the difference between unadjusted raw values in GE and the age/sex-based predicted values from the gene-non-expanded (GNE) model. The horizontal black dashed line across YTO (x-axis) represents the GNE control participants. The YTO horizontal axis is inverted for visualization purposes.
Mixed-effects regression models were used to assess YTO change in GE subjects, controlling for age-related growth derived from GNE controls (indicated by dashed line). Figure 4 shows the results for the functional measures of cognition, behavior, and motor function. Cognitive performance, as measured by the General Abilities Index (GAI), a proxy for IQ, was significantly higher in GE subjects than in GNE controls decades from onset. Behavioral ratings were also superior (indicating less problematic behavior), while motor function differences did not reach statistical significance.
Figure 5 shows the results of the brain volume models. GE subjects exhibited larger total brain, cortical gray matter, and white matter volumes than GNE controls, evident and with the largest group difference at the furthest epoch of time, >50 YTO. These volumes gradually declined, crossing the GNE average around 20 YTO, and became significantly lower than controls within 10 YTO. In contrast, striatal and pallidal volumes remained comparable to GNE until ∼20 YTO, after which they underwent a steep and accelerated decline, consistent with early vulnerability of the basal ganglia.
Figure 6 displays the results of the cortex morphology measures. Early advantages in surface area, folding, and curvature, rather than cortical thickness, accounted for the increased cortical volume in GE subjects. Importantly, larger structural measures were positively correlated with GAI, indicating that the observed brain enlargement is directly related to functional advantage rather than pathological overgrowth which can be seen in some developmental disorders such as autism. 15
The Kids-HD dataset represents the widest premanifest range ever evaluated in HD and provides compelling evidence that decades before motor onset, mHTT influences neurodevelopment in an advantageous way, specifically, through enhanced cognitive function due to cortical surface area expansion.
Taken together with earlier work showing that increasing CAG repeat length below disease threshold is associated with greater cortical volume and cognitive performance, the results support a continuum model: each additional CAG repeat confers progressive alterations in huntingtin protein conformation and function, producing a spectrum of effects across the full CAG range (10–59 repeats).
Antagonistic pleiotropy
Although the concept that neurodevelopment plays a significant role in Huntington's Disease (HD) pathoetiology, knitting together exactly how changes in brain development may lead to later life degeneration has not been clearly defined. The studies that report developmental aberration point to the concept of abnormal circuits that are compensated for, but vulnerable to later degeneration. 2 However, the exact mechanism by which development and degeneration are linked remains unidentified. Given the Kid-HD findings of early brain advantage, theories that connect ability to liability are attractive.
Pleiotropy, in genetics, is defined as the expression of multiple traits by a single gene. Antagonistic Pleiotropy (AP) is therefore defined as a phenomenon in which a pleiotropic gene has effects that are antagonistic with beneficial traits paired with detrimental ones – ability and liability. The AP theory is a leading theory of aging or senescence and posits that genes which initially increase the odds of successful reproduction early in life by improving individual fitness eventually lead to detrimental effects of degeneration. 16
The application of AP to HD was first proposed in a theoretical paper by Albin in 1993. 17 Early proponents suggested that the advantage conferred by mHTT may lie in increased fecundity or reduced cancer risk.18–21 One study did report higher fertility rates in HD families, 18 and three studies observed lower cancer incidence,22–24 though a large recent analysis from the ENROLL-HD database did not replicate the cancer finding. 25
A more compelling and disease-relevant application of AP in HD focuses on brain-specific advantages. Given that HD is a neurodegenerative disorder, it is most parsimonious to hypothesize that mHTT exerts beneficial effects on brain development that later transition into vulnerability. Supporting evidence beyond Kids-HD includes a study by Saft et al. reporting enhanced speech performance in preHD individuals. 26 Similar patterns have been observed in carriers of GRN and MAPT mutations (causing frontotemporal dementia), where higher brain volumes and cognitive advantages were found decades before symptom onset. 27
Importantly, AP is grounded in evolutionary theory. Genes that enhance early-life brain function may have conferred selective advantages in human evolution by improving reproductive fitness and survival. Because their deleterious effects manifest only after reproductive years, these genes escape negative selection.16,28 Thus, understanding the role of HTT in human brain evolution may provide critical insight into its dual role as a driver of both neurodevelopment and neurodegeneration.
Linking evolution to AP and HD
Evolutionary biologists have long speculated that simple sequence repeats (SSRs), such as the CAG repeat in HTT, may play an important role in driving phenotypic variability and evolution. SSRs have been implicated in modulating brain development and function in various species, and these dynamic, mutable elements may act as key genetic modulators, particularly in the context of human brain evolution29,30
Work by Cattaneo and colleagues has demonstrated that HTT is a highly conserved gene, and comparative genomic studies suggest that the CAG repeat within HTT has undergone positive selection, with longer repeats observed in more advanced species. Notably, humans exhibit the longest CAG repeat lengths.31–34 In a stem cell model by Cattaneo, increasing CAG repeat length was associated with enhanced neurogenic potential, supporting the hypothesis that longer repeats confer functional advantages during development, contributing to the evolution of a more complex and adaptive nervous system. 35
At the molecular level, these phenotypic gains may stem from structural changes in the huntingtin protein. Polyglutamine (polyQ) tracts, such as the one encoded by the HTT CAG repeat, can stabilize protein-protein interactions through conformational changes that bring protein domains into optimal spatial proximity.1,36 In HTT specifically, increasing polyQ length may enhance the flexibility of an internal domain, facilitating intramolecular proximity and potentially improving protein function with each additional repeat. 37
As illustrated in Figure 7, increasing CAG repeat length may lead to optimized protein conformation, which in turn promotes greater cortical volume and cognitive function. This expanded phenotypic range enhances variability subject to natural selection, enabling species-level evolutionary gains.
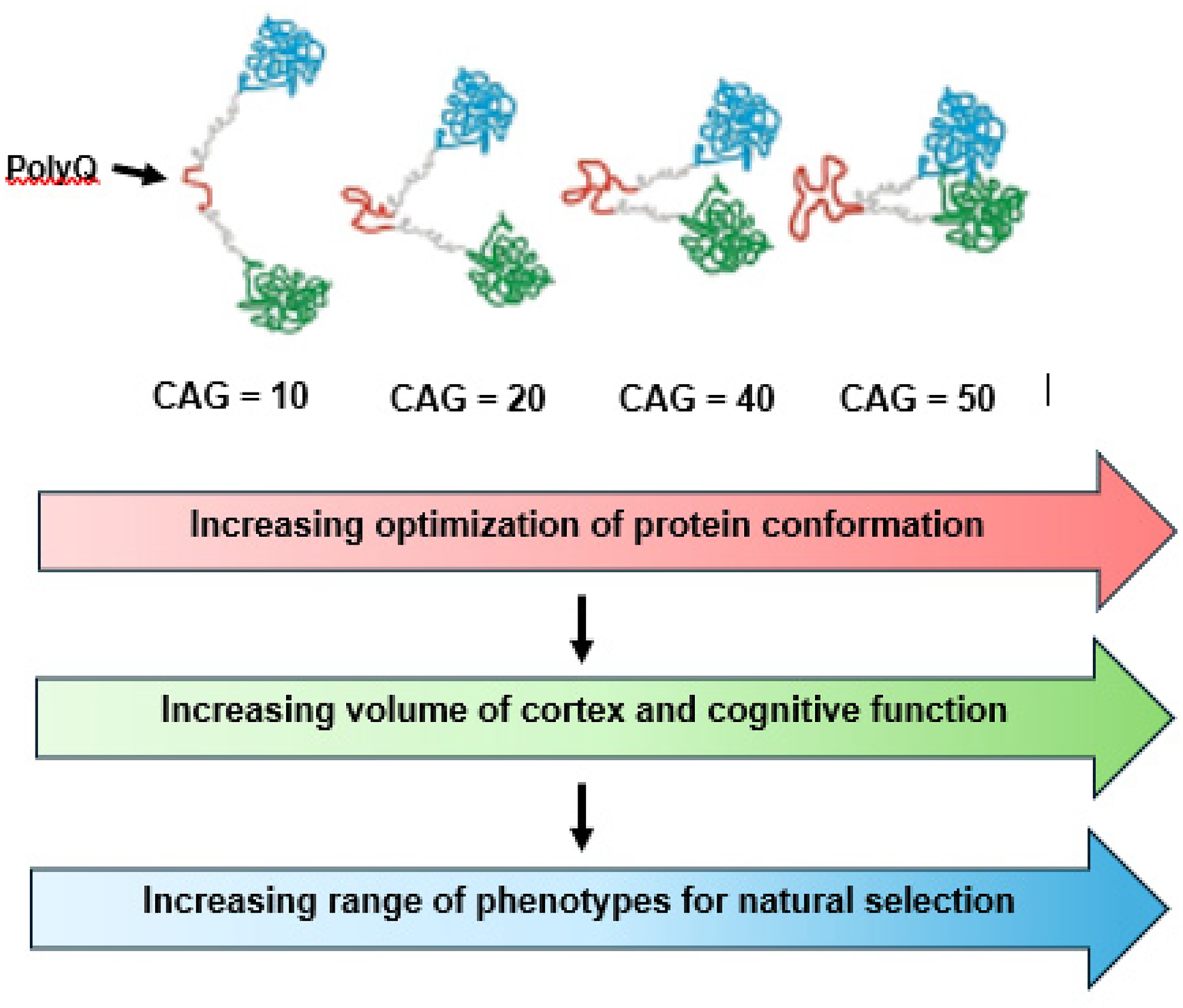
Proposed mechanism for how HTT and mHTT create variations in brain structure and function in the human brain.
Further support for the link between HTT and human brain evolution comes from Kids-HD findings, which showed that increases in brain volume among GE individuals were particularly notable in cortical morphology measures: surface area, folding index, and curvature index. These metrics reflect well-established evolutionary patterns in which the cortex expanded disproportionately, relative to the basal ganglia, during hominid evolution. 38 Importantly, increased surface area (rather than thickness) implies an increase in the number of cortical columns, a hallmark of advanced neural processing and higher intelligence. 39
A recent comparative neuroimaging study found that in humans, but not in chimpanzees, age-related atrophy was most pronounced in cortical regions that expanded during evolution, suggesting that the cost of cortical expansion is increased vulnerability with aging, a hallmark of antagonistic pleiotropy. 40 This idea is further supported by work from Esteves-Fraga et al., who demonstrated that areas of cortical degeneration in HD subjects strongly co-localized with regions showing high expression of developmental genes. 41 This suggests that the very genes driving early brain development, including HTT, may contribute to later neuronal vulnerability and cell loss.
While cortical expansion has been a defining feature of human brain evolution, HD pathology, particularly in its early stages, is characterized by selective degeneration of the striatum. This raises a critical question: How might an expanded cortex contribute to striatal vulnerability? Understanding this relationship may be key to elucidating the full trajectory of HD pathogenesis within an evolutionary and pleiotropic framework.
Mechanisms of linking development to degeneration
A compelling mechanism connecting early cortical advantage to later striatal vulnerability in HD is glutamate excitotoxicity. Glutamate is the brain's principal excitatory neurotransmitter, and the excitotoxicity hypothesis has long been implicated in various neurodegenerative disorders.42–45 In HD, the theory proposes that medium spiny neurons (MSNs) in the striatum degenerate due to sustained glutamatergic overstimulation. Elevated glutamate levels lead to chronic receptor activation, excessive calcium influx, oxidative stress, and ultimately cell death. Recent evidence from an ovine HD model using single-nucleus RNA-seq supports this theory, identifying an MSN-specific glutamate excitotoxicity signature before neuronal death occurs. 46
In the Kids-HD study, GE subjects showed enlarged cortex volumes decades from motor onset. To explore whether this reflected regional effects, we performed a vertex-wise analysis in a subset of participants >20 years from predicted onset (n = 97). As shown in Figure 8, significant cortical enlargements were observed in the sensorimotor, superior parietal, superior frontal, and anterior cingulate cortices—regions known to have dense glutamatergic projections to the striatum. 47 These findings suggest that early cortical expansion may establish an increased excitatory burden on the striatum, potentially priming it for excitotoxic degeneration later in life.
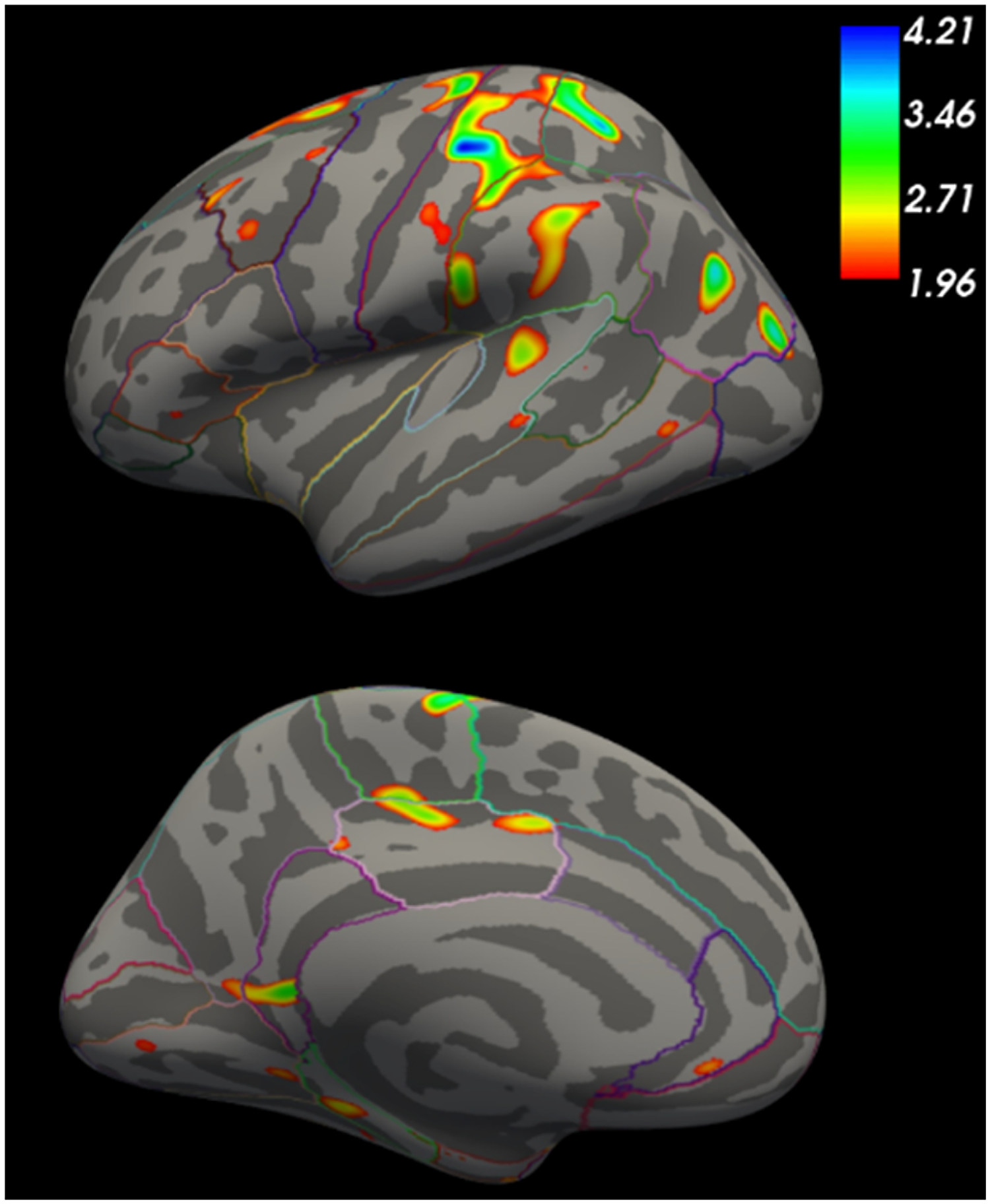
Ge subjects >20 YTO showed cortical enlargement in the sensorimotor and superior frontal cortex.
An important contrast to the ovine model and human studies are the mouse model studies of cortex development that do not support changes that result in advantage, but instead quite uniformly show aberrant cortical development. This is reviewed in the current special edition by Cepeda, Barry and Holley who report that mHTT affects corticogenesis, cell migration and differentiation leading to malformation and hyperexcitability. Although these studies may not seem to support a model of early brain structure and function advantage, there is a rich and deep literature supporting the concept of glutamate toxicity being of primary importance to degeneration including an important early study by Beal et al. 48 and a body of work by the Raymond lab.49–51 Moreover, there are multiple electrophysiologic studies showing abnormal glutamate drive in a variety of mouse models reviewed by Cepeda and Levine. 52 Although there is no consistent evidence to support a developmental increase of cortical glutamatergic inputs to the striatum, the body of literature clearly identifies glutamate toxicity as a primary mechanism of degeneration in HD.
Importantly, glutamate excitotoxicity may trigger a cascade of metabolic stressors, including mitochondrial dysfunction, oxidative stress, and impaired energy metabolism. These disturbances are recognized contributors to both normal aging and pathological neurodegeneration.
Accelerated aging and the AP hypothesis
In aging biology, antagonistic pleiotropy is often framed within the Disposable Soma Theory proposed by Thomas Kirkwood in 1977. This theory posits a trade-off: evolutionary pressures prioritize growth and reproduction over long-term somatic maintenance. 53 As a result, cellular maintenance systems—such as those protecting against mitochondrial damage, oxidative stress, and DNA replication errors—are under-resourced. Therefore, it is these failing cellular maintenance systems that are the mechanisms of the normal aging process. 54
Applying this to HD, one could hypothesize that mHTT-driven cortical expansion imposes excessive metabolic demands, diverting energy away from cellular repair mechanisms. Notably, prefrontal and associative cortical regions, which govern higher-order cognition, have up to 67% higher energy demands than primary sensory-motor areas. 55
In support of the role of energetics, work by Marcy McDonald's lab has shown that HTT has a pleiotropic effect on energy. They used human lymphoblastoid cell lines with repeats 9–34 and showed that increasing CAG repeats resulted in increasing ATP levels, suggesting that HTT plays an important role in cellular energy status. 56 In addition, PET studies of presymptomatic HD subjects shows areas of cortical and subcortical hypermetabolism.57,58 Taken together, these studies support the notion that HTT drives the development of a vulnerable system: as the cortex develops in an energy-intensive fashion, somatic maintenance may be compromised.
When glutamate excitotoxicity begins, the brain's diminished repair capacity may fail catastrophically, accelerating the aging process. A recent review by Machiela and Southwell describes how aging-related cellular processes, such as DNA repair and proteostasis, decline more rapidly in HD models and patients, supporting the theory of accelerated aging as a pathologic mechanism for HD neurodegeneration. 59
Somatic instability and AP
A landmark in HD research is the discovery that somatic CAG repeat expansion in neurons is driven by dysfunction in DNA mismatch repair (MMR) genes. Variants in PMS1, MLH1, MSH3, PMS2, FAN1, and LIG1 have been implicated as modifiers of disease onset and progression. 60 Although these genes are best known for their roles in repair during replication, DNA repair is also essential during development when cell proliferation is high.
The importance of MMR genes in development is supported by disorders such as Cockayne Syndrome, which results from impaired DNA repair and leads to neurodevelopmental failure and premature aging. 61 Importantly, DNA repair capacity declines with age, 62 and impaired repair is one of the nine hallmarks of aging. 54 Thus, DNA repair genes inherently demonstrate antagonistic pleiotropy, supporting the idea that these same genes may contribute both to early developmental success and later-life degeneration in HD. Whether the MMR genes that promote somatic instability in HD also shape neurodevelopment remains an open but important question.
Back to age trajectories: The case of accelerated growth and maturation followed by accelerated aging
In evolutionary biology, life-history theory outlines trade-offs between developmental rate, reproduction, and longevity. For example, species that grow rapidly tend to reproduce earlier but have shorter lifespans. 63 This “fast-slow continuum” or pace-of-life theory is well documented across mammals, with the ratio of development time to adult lifespan remaining fairly constant (about 1:4).64–66 But could such developmental trade-offs also occur within a species, such as among humans?
Recent work by the Thompson lab using isogenic human embryonic stem cells found that expanded CAG repeats (72) led to accelerated neuronal maturation, including longer neurites and a 1200% faster increase in cell size relative to controls. 67 Gene expression pathways involved in cell growth were also upregulated. However, these same neurons died significantly earlier, highlighting the trade-off between accelerated development and early degeneration.
A parallel pattern is seen in the Kids-HD striatal data. As shown in Figure 9, gene-expanded children exhibit early striatal growth, peaking by age 6, followed by continuous decline. 8 In contrast, GNE children reach striatal volume peak around age 14, reflecting the typical neurodevelopmental curve of peak volume then decline driven by maturation and synaptic pruning. Importantly, GE subjects do not show a higher peak volume, but they reach the peak striatal volume much earlier in life. In addition, in repeats greater than 50, the developmental trajectory is CAG-repeat dependent where higher repeats are associated with an earlier peak volume and a steeper volumetric decline. This trajectory supports a model of accelerated growth coupled with premature decline—the signature of antagonistic pleiotropy. Moreover, it highlights again a CAG-dependent process where higher repeats suggest faster maturation and faster degeneration. This is supported by previous reports from the PREDICT study68,69 and our work in subjects with JOHD who show substantially steeper rates of striatal atrophy compared to those with adult onset HD paper. 70

Age trajectories of striatal volume.
Accelerated maturation or early degeneration?
In both the Kids-HD and YAS studies, biomarkers of neurodegeneration (e.g., neurofilament light chain, NfL) emerge ∼20 years before onset7,71 However, in YTO models, cortical volume decline is evident even earlier, beginning as early as 50 YTO and continuing for the next few decades. 14 In those same GE subjects whose cortex was declining in volume between 50 and 20 YTO, there was no NfL increases, suggesting that the early volume loss is not degenerative but may instead reflect accelerated maturation—specifically, synaptic pruning.
Synaptic pruning is a programmed developmental process mediated by microglia and the complement system, known to occur earlier and more aggressively in some neurodevelopmental conditions, including schizophrenia. 72 Thus, early volume loss in GE youth may reflect a precocious or accelerated maturational process. Moreover, this process is normal physiologic activity rather than pathologic degeneration.
Summary: Accelerated growth and maturation (ability) paired with accelerated aging (liability) across the CAG repeat Spectrum
This review integrates concepts from evolutionary biology, developmental neuroscience, and aging to propose a new model for HD pathogenesis. We suggest that mHTT initially promotes a developmental advantage (i.e., a larger, more metabolically active cortex) reflecting its evolutionary role in shaping the human brain. However, this early “ability” is paired with a “liability": the very features that enhance brain performance may also predispose to glutamate excitotoxicity, energy imbalance, and cellular degeneration.
Figure 10 illustrates this framework, showing how HTT accelerates both neurodevelopment and neurodegeneration in a CAG-dependent fashion with increasing repeats resulting in the fastest maturation paired with the steepest degenerative decline.
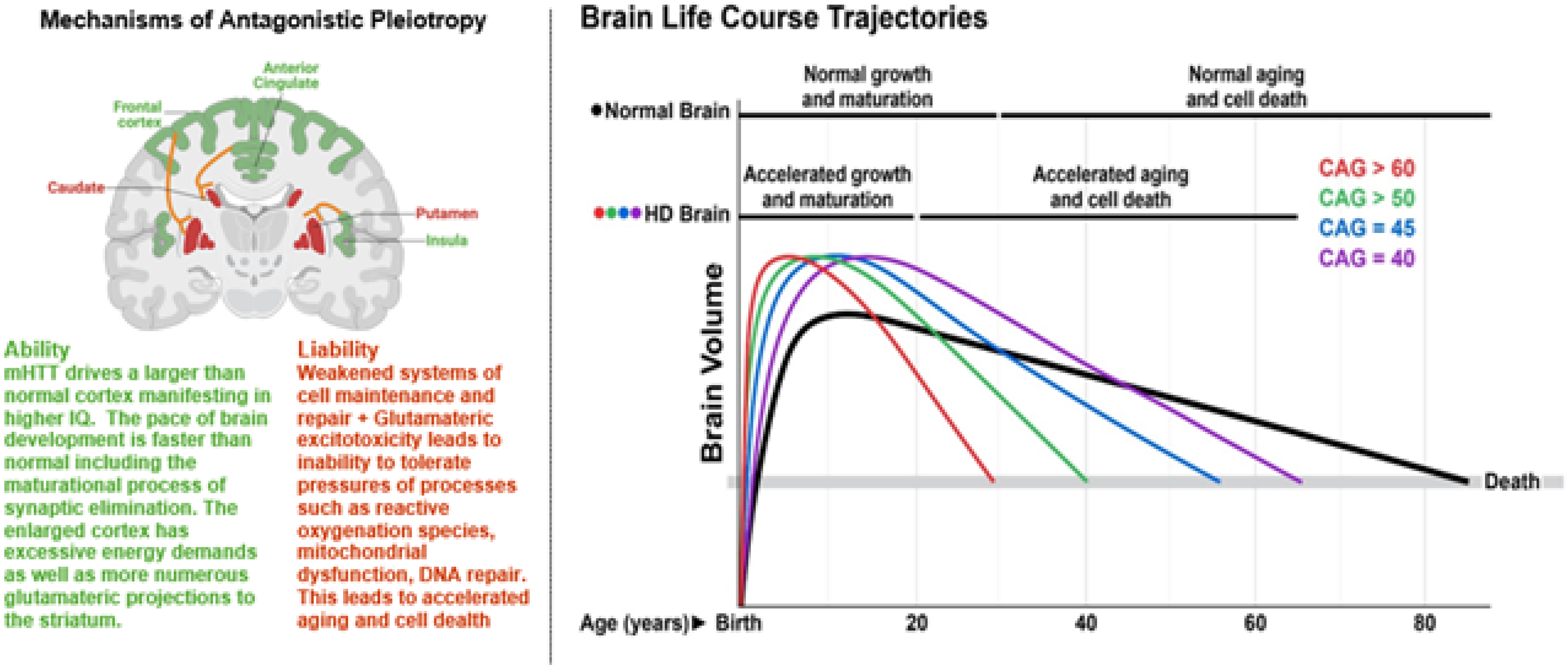
Theoretical concepts of the pathoetiology of Huntington's disease based on antagonistic pleiotropy.
Implications for treatment
This theory not only reconciles conflicting observations across HD research but also highlights novel avenues for understanding and intervening in the disease process. Importantly, this theory challenges the notion that the pathophysiology of HD is solely driven by toxic gain-of-function from mHTT. Instead, it supports the notion that early in life, mHTT produces an advantageous gain-of-function. However, in true ability/liability fashion, mHTT may confer early advantageous gain of function that gives way to toxicity later in life. Given that mHTT may exert its beneficial effects during critical windows of early brain development, lowering it within the first year of life (the timeframe in which it is likely exerting its effect on the cortex) may not only be impractical but could also be harmful. Given that development of the human brain takes a full 30 years, lowering mHTT at any time prior to age 30 could have implications for development. Conversely, targeting mHTT later in life may be insufficient to halt neurodegeneration, as accelerated aging processes may already be underway. A more effective therapeutic strategy may involve a combination of drugs that may target the aging processes in combination with lowering mHTT. As an example, our group recently published data from ENROLL showing that the use of beta-blocker medications both delayed the motor onset of disease and slowed disease progression. 73 What is the mechanism for this? One of the 9 hallmarks of aging is altered intercellular communications leading to impaired tissue function. 54 In HD, the autonomic nervous system (ANS) has been shown to be dysregulated, even in children at risk. 74 This dysregulation causes enhanced norepinephrine release from sympathetic neurons. In the cerebrovascular unit, excessive norepinephrine exposure in endothelial cells may make them desensitized to the production of nitric oxide. Beta-blockers may block the action of norepinephrine in the neurovascular unit and help to maintain nitric oxide production and stabilize normal cerebrovascular function. This is an excellent example of the type of treatment that, paired with a mHTT lowering drug, may work synergistically to maximize neuroprotection.
Footnotes
Ethical statement
The Kids-HD study received approval by the University of Iowa Investigational Review Board (IRB). All participants provided consent prior to study participation.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Kids-HD study was funded by the National Institute of Neurological Disorders and Stroke (R01 NS055903) and the CHDI Foundation (071108). The MRI equipment used in this study was funded by the National Institutes of Health (1S10OD025025-01).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.