
Abstract
Approximate multi-pattern matching is an important issue that is widely and frequently utilized, when the pattern contains variable-length wildcards. In this paper, two suffix array-based algorithms have been proposed to solve this problem. Suffix array is an efficient data structure for exact string matching in existing studies, as well as for approximate pattern matching and multi-pattern matching. An algorithm called MMSA-S is for the short exact characters in a pattern by dynamic programming, while another algorithm called MMSA-L deals with the long exact characters by the edit distance method. Experimental results of Pizza & Chili corpus demonstrate that these two newly proposed algorithms, in most cases, are more time-efficient than the state-of-the-art comparison algorithms.
Introduction
Research on pattern matching is an essential topic in many fields such as big data, knowledge graph, text mining, bioinformatics, and information retrieval. However, compared to exact pattern matching, approximate pattern matching has a wider range of applications. Some examples are finding patterns in a given text to detect text similarity in plagiarism detection [1] and text indexing [2], as well as finding subsequences in a given DNA sequence to detect mutations in bioinformatics [3], etc. Approximate pattern matching refers to finding a pattern in a given text that allows a finite or infinite integer number of errors in the pattern [4]. The error, also known as gap [5], does not care characters [6] or wildcards [7]. In this paper, the error is called wildcards, and the length of wildcards is variable but limited.
There are three possibilities for the problem of wildcards and pattern matching. (1) Fixed-length wildcards, e.g., a***b represents that the characters ‘a’ and ‘b’ only have three errors. (2) Variable-length wildcards, e.g., a* [2, 3] b indicates that the characters ‘a’ and ‘b’ have two or three errors. (3) Wildcards of arbitrary length. It presents that the length of errors can be infinite, e.g., a*[2,
Some exact multi-pattern matching methods have been proposed based on the classic exact single-pattern matching algorithm [8, 9, 10, 11]. However, these methods are simply extended from single-pattern matching. For example,
Two efficient algorithms MMSA-S and MMSA-L based on the suffix array data structure have been proposed [15]. Once the given sequence remains unchanged, the suffix array can be reused. The problem lies in how to achieve approximate pattern matching with wildcards in the suffix array. According to the length of exact characters in the pattern, two algorithms have been respectively proposed by the dynamic programming method and the edit-distance method. When the length is short, the MMSA-S algorithm searches for possible suffixes by the dynamic programming method, and when the length is long, the MMSA-L algorithm computes the possible position intervals by the edit-distance method. Combined with the properties of the suffix array, a large number of impossible positions are excluded, which greatly improves the efficiency of these two algorithms. Based on the experimental results in DNA and Protein sequences, the MMSA-S and MMSA-L algorithms are superior to comparison algorithms in most cases.
The contribution of this work consists of two aspects:
To design and implement two efficient MMSA algorithms for multi-pattern matching with variable wildcards. These algorithms are evaluated experimentally in the dataset of Pizza & Chili corpus. Moreover, these algorithms are more efficient than comparison algorithms. To extend the application of the suffix array in approximate multi-pattern matching. It takes advantage of the suffix array when the order of the objects is constant, and improves the algorithm of suffix array to get the longest common prefix (LCP) array and rank array, which improves the research efficiency.
The structure of this paper is as follows. In Section 2, the related work of multi-pattern matching and suffix array are described. In Section 3, the problem of multi-pattern matching with variable-length wildcards is defined, and some preliminaries of suffix arrays are introduced. In Section 4, our algorithms for multi-pattern matching with variable wildcards based on suffix array are illustrated with some pseudo-codes and figures. Section 5 discusses the experimental and analytical results of the two proposed algorithms compared to other algorithms. Section 6 summarizes the challenges and future research issues.
This section introduces the related work of pattern matching with wildcards, approximate multi-pattern matching, and the applications of suffix arrays and suffix trees in multi-pattern matching or approximate pattern matching.
The problem of pattern matching with wildcards has been studied by many researchers. Fischer et al. [7] first considered the problem of single pattern matching with wildcards or gaps in 1974. However, in their research, the range of wildcards was fixed. In later studies, many methods were applied to single pattern matching with wildcards, gaps or unrelated characters, such as dynamic programming [16], Fast Fourier Transform (FFT) [17] and bit parallelism [18]. In 2002, the pattern matching algorithm with constant wildcards was put forward by Cole et al. [19]. The time complexity was O(nlogn). In 2007, Zhu et al. [20] proposed the algorithm for pattern matching with flexible wildcards. In 2011, Haapasalo et al. [21] proposed an algorithm based on the Ahot-Corasick automaton method to solve pattern matching with arbitrary length wildcards. In 2012, Bille et al. [22] studied an algorithm for finding certain substring in a given text, and the ending position of the substrings was fixed. In these studies, the length of wildcards varied from fixed to variable, or even infinite. Furthermore, these studies focused on the problem of single-pattern matching with wildcards.
However, there are few studies on the problem of multi-pattern matching with wildcards, especially when the length of wildcards is variable. Based on the classic exact single-pattern matching algorithms, some exact multi-pattern matching methods have been proposed, such as MultiBDM [23] and Dawg-Match [24]. However, compared with the exact multi-pattern matching, approximate multi-pattern matching has been less studied, and its research problem is more complicated [25]. In 1996, Muth et al. [26] proposed the MultiHash algorithm for multi-pattern matching with one error. In 1997, Baeza-Yates et al. [27] designed an extension of PEX algorithm, known as MultiPEX algorithm, which matched the exact pieces splitting the patterns. These algorithms were not specific to the problem of multi-pattern matching with wildcards. In 2007, the TARA algorithm [28] was proposed by bit parallelism for the fixed-length wildcard in multi-pattern matching. Nonetheless, the method of bit parallelism is efficient only when the length of the pattern is small. In 2011, three sub-algorithms based on the method of Fast Fourier Transforms (FFT) were proposed by Zhang et al. [29] for multi-pattern matching with fixed wildcards. These algorithms are applicable even if the number of patterns is small. In 2018, Biswas et al. [30] provided a linear spatial index of the ranked document retrieval for multiple-pattern queries through theoretical analysis.
Suffix array was constructed based on the trie and suffix tree data structures. The trie was derived from the word of retrieval and was proposed by Edward Fredkin [31] in 1960. It is a variant of the Hash tree and stores all sub-strings in an ordered tree structure. Its retrieval efficiency is higher than that of the Hash tree. In 1973, Weiner [32] first proposed a new data structure suffix tree based on the trie structure, which stores all the suffixes of a given sequence in the tree structure, and the path from the root node to the leaf node is a suffix. As an efficient and popular data structure in exact pattern matching, suffix tree was then used for string retrieval, string matching, and natural language processing, etc. However, few existing studies focus on the pattern matching with wildcards based on suffix tree. In 2005, the inexact-suffix tree was introduced by Chattaraj et al. [33] to detect the extensible patterns. However, the time complexity of this algorithm was very large. In 2009, Ukkonen [34] introduced an approach based on suffix tree algorithm to find the equivalent representation in gapped or non-gaped motifs. In 2014, Bille [35] put forward two methods to deal with the problem of pattern matching with wildcards through the suffix tree. They constructed a suffix tree to store all suffixes, including all possible positions of wildcards. However, these methods were described theoretically. Time complexity increased with the number of wildcards in the pattern. Therefore, these methods mentioned above are difficult to implement.
Similar to suffix tree, the suffix array has more achievements in the single-pattern matching and exact pattern matching than that in the multi-pattern matching and approximate pattern matching. In 2006, Rahman et al. [36] presented an algorithm to find patterns with variable length gaps through the suffix array method. However, the algorithm was just a theoretical description, and the pattern had fixed length gaps. In 2014, Shrestha et al. [37] not only provided a detailed description of SA-IS algorithm [38], but also described techniques that suffix arrays adapt to inexact matching in biological application. In this work, it directly supports the exact ‘subset’ matching by adopting the lexical ordering of suffixes in the suffix array. Some subset matchings allow certain characters to be ignored in predefined position-specific way by the edit-distance method. However, this work can only be applied to genomic sequence with short inexact length. In 2016, Thankachan et al. [39] proposed a suffix model to cope with the approximate sequence matching problem involving a selected bounded set of perturbed suffixes. However, this method was limited to theoretical analysis and lacked practical demonstration. In 2017, Hon et al. [40] studied two dictionary matching problems with one gap and one missing substring based on the index data structures, such as suffix trees and suffix arrays. They had theoretically described the solution in terms of space efficiency and succinct space. In 2018, two algorithms for multi-pattern matching with variable-length wildcards based on the suffix tree were presented in our previous work [41].
Inheriting the advantages of index suffix data structures, two new algorithms based on suffix array have been proposed, which are more efficient and wildly applicable than the method based on suffix tree, especially when all suffixes are sorted in lexicographic order.
Preliminaries
In this section, some necessary background information will be presented for the understanding of the new algorithm MMSA (Multi-pattern Matching based on Suffix Array), which involves techniques related to the pattern matching with wildcards, the multi-pattern matching, and the suffix array.
Pattern matching with wildcards
The basic problem of pattern matching with wildcards is to find all possible positions that match the exact characters of the pattern and satisfy the range constraints of wildcards in the object sequence. Let the pattern
Pattern matching with fixed-length wildcards. When 0
Pattern matching with periodic-length wildcards. When 0
Pattern matching with arbitrary-length wildcards. When 0
Pattern matching with variable-length wildcards. When 0
where 1
An example of pattern matching with wildcards.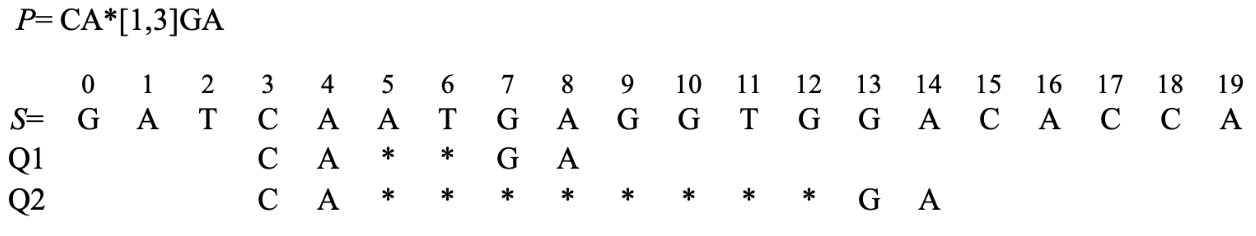
For the first character group CA, the starting positions are 3, 15, 18, respectively; for the second character group GA, the starting positions are 0, 7, 13, respectively; the minimum value of wildcards is 1 while the maximum value is 3.
Multi-pattern matching with wildcards is more complicated than single-pattern matching with wildcards. The following relationships between patterns and matching methods are required to be considered.
To satisfy all patterns; To satisfy any pattern; To satisfy pattern A but not pattern B; To match one pattern at a time; To match multiple patterns simultaneously;
This work only considers the problem of matching each pattern with variable-length wildcards respectively at one time, and the output of any pattern can be satisfied.
An example of multi-pattern matching with wildcards.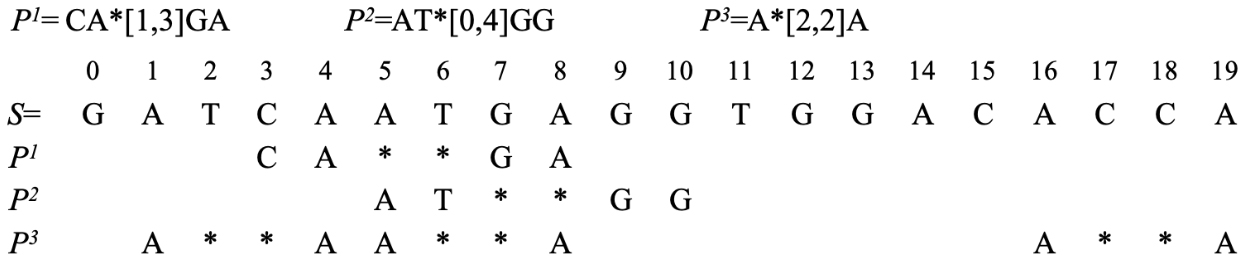
According to Example 1, the satisfying matching occurrence of pattern
Let
Suffix array
According to Manber et.al [15], the suffix array is an array in lexicographic order. Let
An example of the suffix tree and suffix array for a sequence.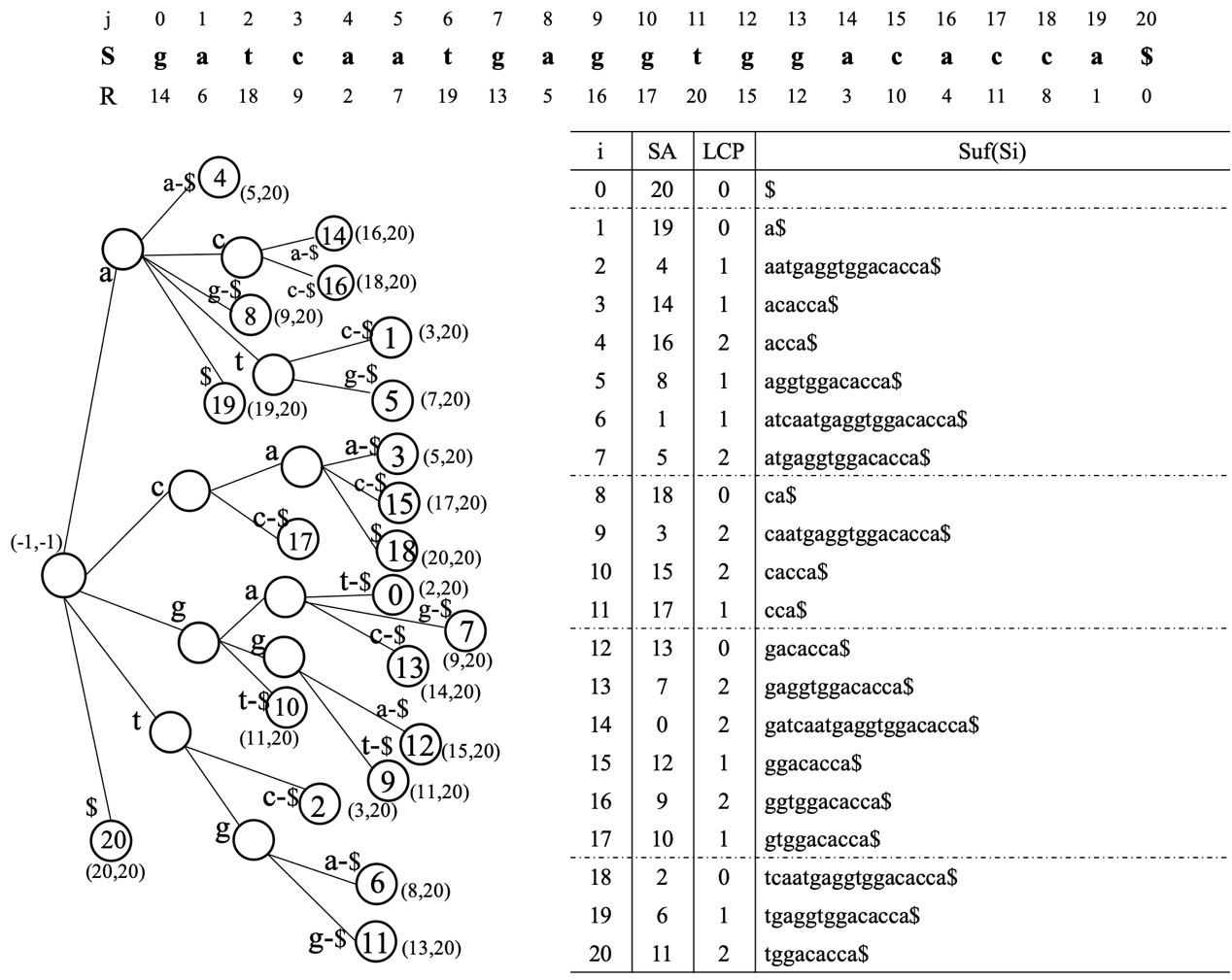
Compared with the suffix tree, all suffixes are stored lexically in the suffix array. For example, the suffix ‘a$’ stores the starting position of the suffix, so SA[1]
In addition, LCP[4]
This section introduces the idea of the algorithm MMSA for multi-pattern matching with variable-length wildcards based on suffix array. First, this study gives the construction and pre-processing steps of the suffix array. The suffix array is constructed according to the algorithm SA-IS of Nong’s method [38], in which the rank array and the longest common prefix array are added. Subsequently, the two algorithms, MMSA-S and MMSA-L, are designed according to the length of the pattern. Finally, the complexities of the two algorithms are analyzed.
Suffix array construction
Different from the suffix tree, the suffix array is not simply built according to the suffix tree from the root to leaves by order. Several algorithms are presented in linear time. The time and space complexities of these constructing algorithms vary with the sorting method. In this paper, the algorithm MMSA is based on the suffix array constructing algorithm SA-IS by Nong Ge et al. [38]. Induced sorting is applied to SA-IS algorithm. According to some properties, the character
According to the requirement of this work, the rank array and the longest common prefix (LCP) array are added on the basis of the SA-IS algorithm. The improved algorithm is given in the form of Algorithm 1.
When the object sequence
Pre-processing
In order to improve the properties of the suffix array in our algorithm, two sub-algorithms have been proposed according to the length of the exact characters in the pattern. The pre-processing steps are different in the two algorithms.
For the MMSA-S algorithm, the length of exact characters in a pattern is short, e.g. a*[0, 3] g, t*[1, 2]cc*[0, 1] a. In the pre-processing phase, it needs to sort all patterns alphabetically according to the first exact character. In addition, it computes the maximum and minimum length of each pattern. According to Definitioms 1 and 2, this pre-processing method will improve the efficiency of multi-pattern matching in the MMSA-S algorithm.
For the MMSA-L algorithm, the length of exact characters in a pattern is long, e.g. aggt *[1,2] ccat, gagat*[1,3] ttag*[0,2] cccg. In the pre-processing step, all exact characters need to be divided into different groups by the wildcards. The groups of all these characters are then sorted alphabetically. If these groups have a common prefix, it will simplify the algorithm MMSA-L. When the pattern is matching, it starts with the last character group of each pattern. There will be no match if the last character group does not exist. By combining the maximum and minimum length of each pattern, some positions that do not satisfy the matching requirements will be excluded according to Definition 2. This pre-processing technique will avoid some unnecessary steps and improve the efficiency of the MMSA-L algorithm.
The MMSA-S algorithm
The MMSA-S algorithm is suitable for the short exact characters in the pattern. Suppose pattern set
An example of the MMSA-S algorithm.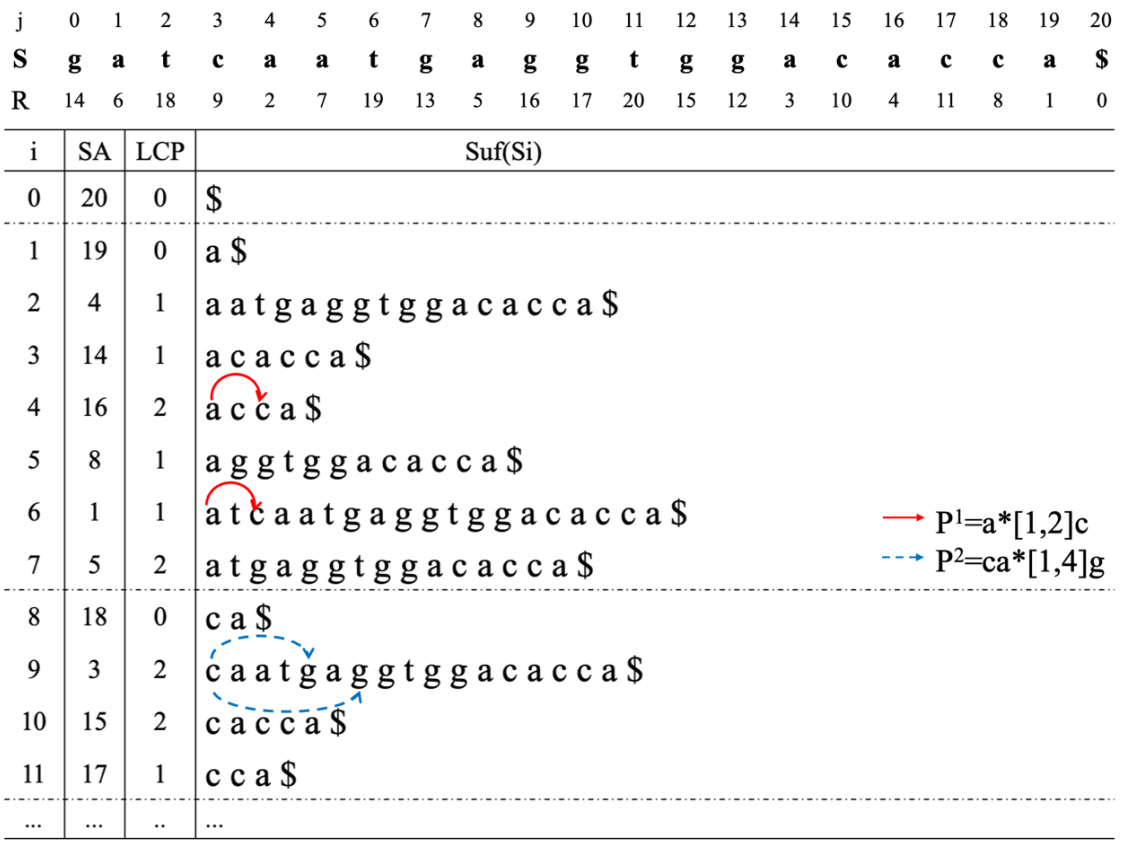
In Fig. 4, an example of suffix array construction is given for the object sequence
Next, the pattern
The MMSA-L algorithm is suitable for the pattern with long exact characters. Suppose pattern set
Figure 5 shows an object sequence
In pattern
This example demonstrates that if the exact character group is longer, the fewer times it will appear. Meanwhile, it eliminates other impossibilities and improves the efficiency of the algorithm.
An example of the MMSA-L algorithm.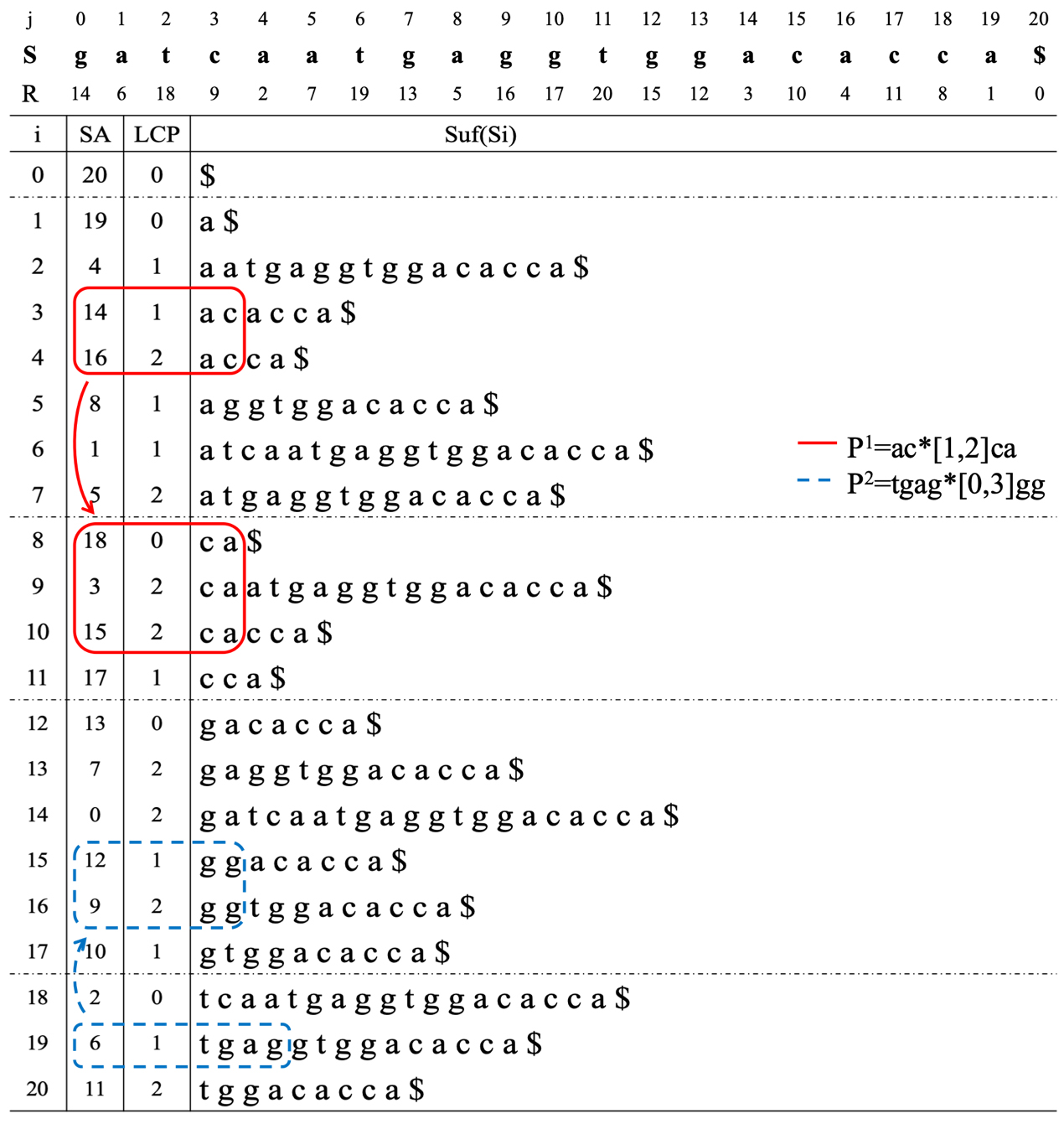
In this section, the complexities of MMSA-S and MMSA-L will be discussed respectively. In the construction of suffix arrays, the SA-IS method is adopted by both algorithms. The time complexity is
For MMSA-S, there is one ‘for’ loop to sort
The MMSA-L algorithm has two ‘for’ loops in the pre-processing phase and two ‘for’ loops in the matching phase. The time complexity of the MMSA-L algorithm is
According to the above analysis, the time complexity is
In addition, the complexities of using suffix tree for multi-pattern matching with our previous work [41] is compared. The time and space complexity of MMST is
Experiments
In this section, the time performances of the MMSA-S and MMSA-L algorithms were compared with other algorithms through experiments, which were performed on a computer with an Intel Core i7-7700 3.6 GHz CPU, 8 G of DDR3 RAM, and running on a Win7 64-bit operation system. All codes were written in Java and compiled with IntelliJ IDEA CE. Comparison algorithms include WM-gap, BG-gap, ST-TWEC-gap, MMST-S and MMST-L. The WM-gap algorithm was reconstructed according to the Wu-Manber algorithm [11] while the BG-gap algorithm was reconstructed according to the Multiple BNDM algorithm [23]. The reconstruction method was proposed from the thoughts of Salmala [43] and Ukkonen [44], while the ST-TWEC-gap algorithm was recomposed based on the T-TWEC algorithm [45].
The test dataset is the Pizza & Chili corpus [46]. The size of the alphabet in natural language is larger than that of DNA sequences and protein sequences. The matching repetition rate of natural language is lower than that of DNA sequences. Therefore, DNA and PROTEINS datasets were used in this work. The length of the DNA sequence is 2,293,760 while the size of the alphabet
Experiments on DNA sequence
Based on the same environment and datasets, performance will be compared when a variable changes. In these experiments, variables include the length of the object sequence, the number of patterns, the length of exact characters in the pattern, and the number of wildcards.
Different length of object sequence
Change the length of the object sequence to 10,000
Comparison of different lengths of DNA sequences.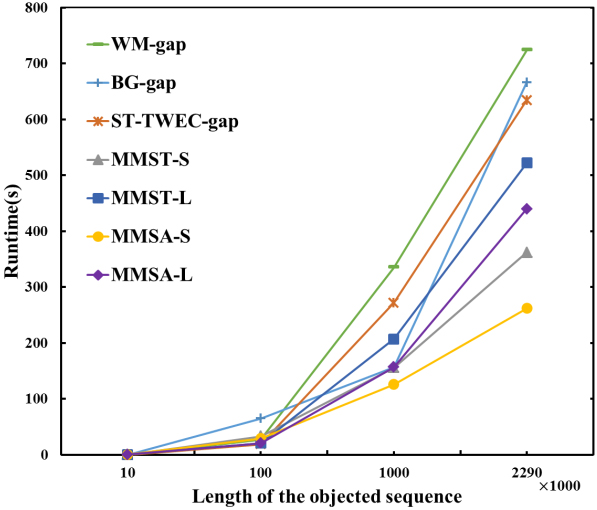
Change the number of patterns
Comparison of different numbers of patterns in DNA sequences.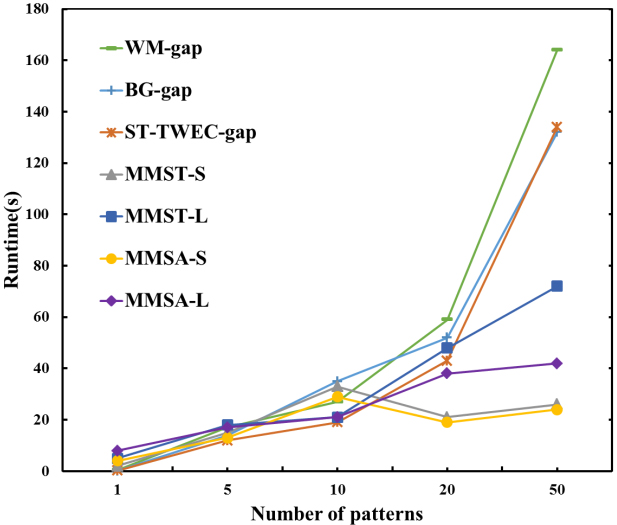
Change the length of exact characters in a pattern
Comparison of different lengths of exact character in DNA sequence.
Change the number of wildcards in each pattern
Comparison of different number of wildcards in DNA sequence.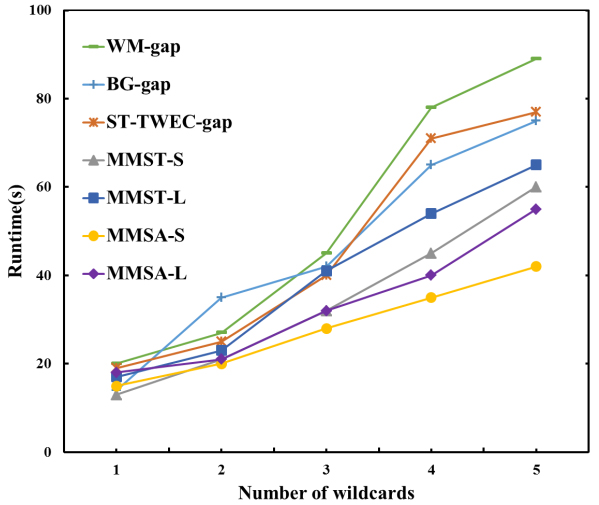
Comparison of different lengths of Protein sequences.
The protein sequence is closer to natural language than the DNA sequence. The alphabet size of the protein sequence is 20, which is 26 in English. Compared to the DNA sequence, the protein sequence will be more efficient in natural language because of its larger alphabet size, fewer repeat positions, lower frequency of satisfied pattern matching, and higher efficiency. In this research, the protein sequences were taken from the PROTEINS datasets of Pizza & Chili corpus. The length of this protein sequence is 1,699,484, which consists of 20 alphabets, such as {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}.
The following four comparative experiments have compared the time performance with four variables in seven algorithms. When another environment is the same, it will change one variable at a time. For example, the length of the object sequence, the number of patterns, the length of the exact character in the pattern, and the number of wildcards.
Different lengths of object sequences
As shown in Fig. 10, the length of the object sequence is changed to 10,000
Different numbers of patterns
In this experiment, the numbers of patterns
Comparison of different numbers of patterns in Protein sequences.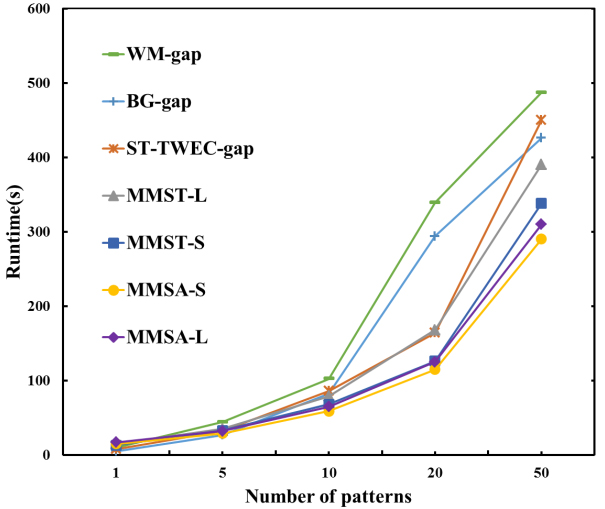
To compare the influence of the exact character length
Comparison of different lengths of exact characters in Protein sequences.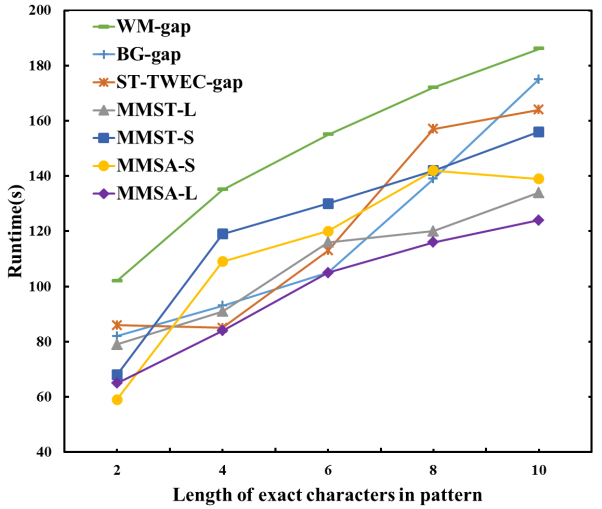
Comparison of different numbers of wildcards in Protein sequences.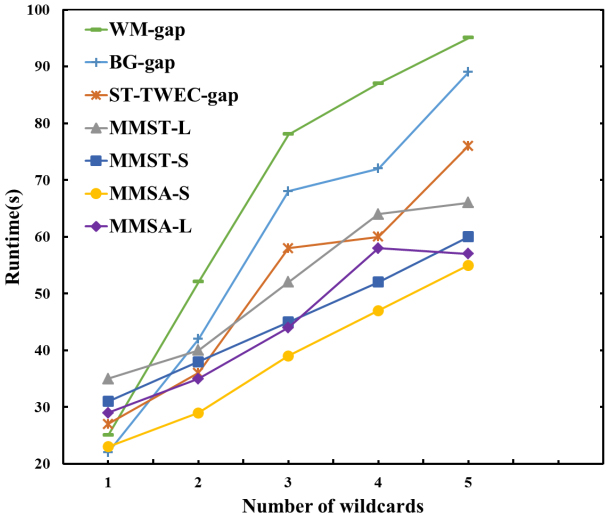
As shown in Fig. 13, the number of wildcards in each pattern
Conclusions and future work
In this paper, two MMSA algorithms based on the suffix array are proposed, which have higher time performances than the other comparison algorithms in multi-pattern matching and approximate pattern matching. According to the properties of the suffix array, all suffixes are sorted alphabetically, and the longest common array is easily available. These attributes can exclude many impossible positions and make the two algorithms more efficient. The MMSA-S algorithm is suitable for the short-exact characters in a pattern by the method of dynamic programming, while the MMSA-L algorithm is applied to the long-exact characters in a pattern by the edit-distance method.
The disadvantages and limitations of these two MMSA algorithms include the following two aspects. Firstly, the suffix array requires more space to store all suffixes than the other comparison algorithms. In addition, it will take some time to construct the suffix array, but these procedures will be very useful in the processes of query and matching. Secondly, in the case of a small number of patterns, the efficiency of the two MMSA algorithms is not as good as that of the other comparison algorithms. These two MMSA algorithms are applicable to a large scale of pattern sets.
This research leaves several problems for further study. One of the important problems is to make full use of the two MMSA algorithms in practical applications. Once the suffix array is constructed for reuse, it is suitable for full-text indexing and full-text searching. In addition, the property of the longest common array in the suffix array is suitable for Chinese word segmentation to eliminate ambiguity in natural language processing. Chinese words can be segmented or disambiguated by taking the frequency of words or phrases in the suffix array as the longest common substring, which will be feasible and more efficient. Therefore, it is our next research issue in the future.
Footnotes
Acknowledgments
This research was supported by the National Key Research and Development Program of China (Grant No. 2016YFB1000900) and the National Natural Science Foundation of China (NSFC) (Grant Nos. 61503116 and 61229301). In addition, we would like to thank the anonymous reviewers who have helped to improve the paper.