
Abstract
Several studies of deep brain stimulation (DBS) of the fornix or the nucleus basalis of Meynert have been recently conducted in people with Alzheimer’s disease, with several recruiting participants <65 and thus have early-onset Alzheimer’s disease (EOAD). Although EOAD accounts for less than 5.5% of AD cases, ethical considerations must still be made when performing DBS trials including these participants since a portion of people with EOAD, especially those possessing autosomal-dominant mutations, have an atypical and more aggressive disease progression. These considerations include appropriate patient selection and signing of an informed consent for genetic testing; appropriate study design; potential outcomes that people with EOAD could expect; and accurate interpretation and balanced discussion of trial results. Finally, recommendations for future DBS for AD trials will be made to ensure that EOAD patients will not experience avoidable harms should they be enrolled in these experimental studies.
Keywords
INTRODUCTION
There has been a recent surge in experimental trials on deep brain stimulation (DBS) for Alzheimer’s disease (AD) [1–5], with several studies recruiting participants <65 [1, 3–5] and thus have early-onset Alzheimer’s disease (EOAD). Although there have already been previous discussions on the ethics of DBS for neurodegenerative disorders [6–9], issues arising from recruiting people with EOAD for DBS trials remain unexamined and unexplored in the literature. To address this gap, we discuss potential ethical issues focusing on selection criteria, genetic testing and informed consent, study design, measured outcomes, and result interpretation and portrayal to protect people with EOAD participating in DBS for AD trials.
EARLY-ONSET ALZHEIMER’S DISEASE AND GENETIC PREDISPOSITION
Dementia affects an estimated 46.8 million people worldwide [10], with AD as its leading cause [11]. People with AD dementia suffer from cognitive or behavioral impairment in two or more domains, which include memory, reasoning and executive function, visuospatial abilities, language functions, and personality, that significantly interferes with ability to function at work or at usual activities [12].
Although the majority of people with AD are ≥65 [13], 5.5% of those affected have an onset of dementia before 65 [14] and thus are classified as having EOAD. The 65-year-old cut-off point has no biological significance and is mainly an indicative of social divide in terms of employment and retirement age [15]. Nonetheless, people with EOAD usually have a more rapid disease progression and have more pronounced brain pathology compared to those who develop AD symptoms after 65 [16]. In addition, they have a much shorter survival time; much more prevalent language disturbance [17]; exhibit other atypical symptoms such as visual agnosia, apraxia, dyscalculia, and executive dysfunction [18]; have a higher prevalence of additional non-cognitive neurological symptoms [19]; and exhibit more severe temporoparietal junction atrophy [20].
At least 62% of patients with EOAD have a history of AD in the family [21], implying potential genetic underpinnings. Currently, three genes have been fully established to cause highly penetrant and autosomal dominant AD: amyloid precursor protein (APP) and presenilin 1 and 2 (PSEN1 and PSEN2). PSEN1 makes up 30 to 70% of familial EOAD (EOFAD); followed by APP that accounts for 10–15% of EOFAD cases; and lastly, by PSEN2 that accounts for less than 5% of all EOFAD [22, 23]. Mutations in APP causes its aberrant processing and increased Aβ42 secretion, whereas mutations in PSEN1 or PSEN2 lead to aberrant cleavage of APP by γ-secretase, resulting in an overproduction of Aβ42 [16]. Overall, this leads to the biological cascade causing the observed cognitive defects in AD. The age of AD onset in PSEN1 mutation carriers is between 30 and 50 years old, 40 to 70 years in PSEN2, and 45 to 60 years in APP mutation carriers. Atypical presentations such as language impairment and behavioral symptoms such as delusion, hallucinations, and apathy have been observed in those with PSEN1 or PSEN2 mutations [22]. Certain APP mutations have also been linked to cases of congophilic angiopathy [23], which can lead to leukoencephalopathy, stroke-like episodes, hemorrhage, and cortical calcification [19].
Even though APP, PSEN1, and PSEN2 mutations are the only ones definitively proven to cause autosomal-dominant EOAD, the presence of an APOE ɛ4 allele has also been associated as a risk factor for typical AD and potentially reduces its age of onset by roughly 10 years. It is not a necessary component though since patients who typically exhibit an atypical and early-onset AD course, exhibiting focal cortical, non-memory impairments, and a more aggressive progression, can develop AD even in the absence of an APOE ɛ4 allele [18]. Nonetheless, patients who have APP, PSEN1, or PSEN2 mutations could also have a much earlier age of onset if they possess an APOE ɛ4 allele [23, 24].
Currently, people diagnosed with EOAD are given the same treatment as those who have late-onset Alzheimer’s disease (LOAD), given similarities in pathogenesis and clinical features [19]. Only six drugs are FDA-approved for the management of AD symptoms; however, none of them stops disease progression [25], treats the underlying pathology, or provides long-term benefit [26]. As such, several clinical trials on different modes of treatment are being undertaken to either provide additional long-lasting relief from symptoms or treat the underlying AD neuropathology. Among the treatment modalities being investigated is DBS, a procedure wherein leads are inserted into the brain region of interest to deliver continuous electrical stimulation [27], with the hope of ameliorating cognitive dysfunction. Currently, DBS has regulatory approval for essential tremor, Parkinson’s disease, dystonia, obsessive-compulsive disorder, and epilepsy [28].
CLINICAL STUDIES ON DEEP BRAIN STIMULATION FOR AD
The first experimental trial on DBS for AD was performed in 1984 where the nucleus basalis of Meynert (NBM) was stimulated. Although there was no improvement in memory or cognition, preserved cortical glucose metabolic activity in the left parietal and left temporal lobes and partial arrest of deterioration in the left frontal area were observed [29]. The next clinical trial was performed 26 years later [1], and it was driven by a serendipitous discovery in 2008 when DBS of the fornix to treat obesity resulted to “deja vu-like” sensations during surgery and improvements in episodic verbal and associative memory after three weeks of stimulation [30]. The 2010 Phase I trial investigated DBS of the fornix in six patients with early AD. Similar to the 2008 study, two patients experienced autobiographical experiential phenomena during surgery. In addition, after 12 months of continuous DBS, some patients were reported to have improved memory and reduced cognitive decline, reversed glucose metabolism [1], and increased hippocampal volume [31]. Given that the Phase I trial was considered to have proven the safety of DBS of the fornix and showed metabolic changes associated with it, a Phase II randomized, double-blind, placebo-controlled, delayed-start trial is currently being conducted in 42 subjects with mild, probable AD. In this trial, half of the subjects will not receive any stimulation while the other half will receive continuous DBS stimulation for 12 months; after which, all participants will receive stimulation for 12 months [32]. Results of the first year of this trial have already been published and indicate no significant difference in cognitive scores between those who received and those who did not receive stimulation. However, stratifying participants based on age showed that those who are <65 actually significantly worsened after DBS for one year, whereas those ≥65 had a slight improvement in cognitive function. In terms of safety, there were 145 and 117 non-serious adverse events in patients that received and did not receive stimulation, respectively. In addition, nine serious adverse events for each participant subgroup were reported. Serious adverse events include those that lead to prolonged hospital stay, new hospital admission, disability, or death, such as infection, lead repositioning, post-op nausea, depression, suicidal ideation, and worsening confusion. Non-serious adverse events are predominantly general medical in nature, followed by psychiatric events. Taking into account the nature and extent of reported adverse events, an independent data and safety monitoring board concluded that the observed safety profile was as expected with deep brain stimulation [3].
Aside from the aforementioned Phase I [1] and Phase II trials [3] of fornix DBS in North America, several other case studies and trials of DBS for patients with AD have been reported. A team in France performed fornix DBS in a patient with mild cognitive decline. After 12 months of stimulation, the patient’s cognitive performance reportedly stabilized, and the patient also had increased mesial temporal lobe metabolism [2]. Aside from the study done in 1984 [29], another trial of DBS of the NBM was also performed by Kuhn et al. [4] where six patients with mild to moderate AD received bilateral DBS. Their study consisted of an initial one-month randomized sham-controlled stimulation phase, where two weeks of stimulation was followed by two weeks without stimulation or vice versa, and a succeeding 11-month phase of continued open stimulation on all patients. During the first month, mean Mini-Mental State Examination (MMSE) scores improved after two-weeks of stimulation compared to the score after two weeks without stimulation. After almost a year of stimulation, cognitive assessments revealed slower disease progression when compared to patients undergoing medication. In addition, some patients exhibited increased temporal and amygdalo-hippocampal glucose metabolism after almost a year of stimulation. In terms of safety, the surgical procedures were well tolerated, and the patients had fast recovery and did not have significant stimulation-induced untoward effects [4]. Kuhn et al. [5] then further extended their study and performed continuous DBS of the NBM in two patients who have an average age younger than the average of those in the Phase I trial and who both have lower baseline ADAS-Cog scores. One participant deteriorated after 26 months based on ADAS-Cog and MMSE scores, whereas the other participant had a stable ADAS-Cog and even improved MMSE score after 28 months. Hardenacke et al. [33] then collated the results of the Phase I trial [4] and that of the two new patients [5] and suggested that NBM-DBS performed at a younger age and at an earlier disease stage may favorably impact cognitive functions and disease progression.
ETHICAL CONSIDERATIONS ON DBS STUDIES ON PEOPLE WITH EOAD
Majority of the trials [1, 32] performed or currently ongoing recruited patients who are less than 65 years old, and thus could potentially have EOAD. Given that DBS is an invasive procedure that could lead to a number of neurologic and psychiatric unwanted side effects [34], it is important to consider ethical issues that may arise when performing it to different patient subgroups, especially to individuals less than 65 who might have certain mutations that could lead to a more aggressive disease course [16].
Considerations for patient selection
Four out of six reported studies [1, 32] posted recruitment details on the clinicaltrials.gov database and described them in their papers (Table 1). From this information, it is evident that five out of six DBS for AD studies recruited a total of 19 participants <65, all of which have at least mild cognitive impairment. This indicates an overrepresentation of EOAD in the study population (32.7%) given that only around 5.5% of people with AD have an early disease onset [14]. Such overrepresentation of EOAD patients might not have been deliberately made by the authors and could have just been a result of better success in recruiting and enrolling younger patients due to their greater capacity to tolerate surgery [35] and provide consent [36]. Furthermore, these studies did not mention performing any family background checks or genetic tests for APP, PSEN1, and PSEN2, which makes it possible that a participant enrolled could also have familial (EOFAD) or autosomal-dominant EOAD (AD-EOAD). If one considers that the proportion of autosomal dominant EOAD patients among all EOAD patients is 13% [21], then it seems likely that at least two patients (13% of 18 = 2.34) with AD-EOAD have already participated in these trials. Interestingly, patient 3 in the Phase I trial of Laxton et al. [1] was below 65, had an aggressive disease course prior to surgery based on MMSE scores, and had the worst outcome post-DBS based on MMSE scores. As such, patient 3 might actually have a form of EOFAD or AD-EOAD; however, this cannot be ascertained given that no family background checks or genetic tests were presented in the report. Considering that people <65 are being recruited in DBS for AD studies, we suggest considerations and adjustments in certain inclusion and exclusion criteria when patients with EOAD are potentially recruited in studies, especially those who have autosomal-dominant EOAD.
Criteria for patient recruitment in studies on DBS for AD and number of recruited participants who are less than 65
NBM, nucleus basalis of Meynert; NINDS-ADRDA, National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Disorders Association (ADRDA); DSM, Diagnostic and Statistical Manual of Mental Disorders; ICD10, World Health Organization International Statistical Classification of Diseases and Related Health Problems; NIA-AA, National Institute on Aging-Alzheimer’s Association; CDR, Clinical Dementia Rating scale; MMSE, Mini-Mental State Exam; FCSRT, Free and Cued Selective Reminding Test; ADAS-Cog 11, Alzheimer’s Disease Assessment Scale – cognitive component; C-SSRS, Columbia Suicide Severity Rating Scale; CSF, cerebrospinal fluid.
First, since certain AD-EOAD patients could exhibit atypical behavioral symptoms and DBS could have unwanted psychiatric effects [34], recruited EOAD patients should not have any major psychiatric disorder, especially those that increase the risk of suicide, such as depression, schizophrenia, and substance use disorders [37, 38]. Participants with a history of and/or who were experiencing suicidal ideations at the time of recruitment should be excluded in trials considering that suicidality is a potential adverse event of DBS [39–41]; patients with AD have an increased risk of committing suicide [42]; and people with certain autosomal AD mutations have a high risk of depression and disinhibition [43]. Of all the studies of DBS for AD, only two studies [4, 32] specifically excluded patients who had previous suicide attempts or who have suicidal ideations. Moreover, should appropriate consent be given to employ genetic testing in patients with a family history of AD, careful counselling should be provided to minimize the risks of increased suicidal ideation from an untoward result. Proper tests and monitoring should then be made to ensure that individuals with positive results do not exhibit any suicidal tendencies or ideations prior to commencing DBS surgery.
Second, potential adjustments in the required cognitive profile and disease stage of recruited EOAD patients should be made to account for its shorter disease duration and more aggressive course, especially in participants who have autosomal-dominant mutations [22]. In past and ongoing studies of DBS for AD, some trials [1, 32] only recruited patients with mild AD, whereas Kuhn et al. [4] also included those with moderate AD. In terms of cognitive profile, two studies [1, 32] recruited patients with a Clinical Dementia Rating (CDR) of up to 1, two studies [1, 4] recruited patients with MMSE as low as 18, and one study [32] recruited patients that have an ADAS-Cog 11 score as high as 26. The inclusion of patients with CDR, MMSE, and ADS-Cog 11 scores that already signify cognitive decline beyond the mild cognitive impairment stage [44] and at the start of the dementia phase warrants serious consideration when patients with EOAD are included in a study, given EOAD’s more aggressive disease course [16]. Considering the initial results of Laxton et al. [1] and Hardenacke et al. [33] showing that patients in an earlier disease stage are more likely to benefit from DBS, there is a need to modify the cognitive status cut-offs for participants <65 participating in DBS for EOAD studies. EOAD patients that have a CDR score >0.5, MMSE score <23, and ADAS-Cog 11 score >18 [45], and possess mutations predisposing them to more aggressive cognitive deterioration [22, 23] should not be included in DBS studies unless more evidence has been gathered regarding the efficacy of DBS in later AD stages. Although the use of most biomarker data as diagnostic tools has not yet been approved clinically, hippocampal volume, tau, and Aβ cerebrospinal fluid levels, and brain activity [46] could also be used in conjunction with cognitive tests to ensure that enrolled EOAD patients are at an early disease stage.
Third, it is important to consider the effect of excluding or only including certain patient subgroups based on participants’ cognitive, genetic, and/or biomarker profile on the study’s external and internal validity and also to determine whether it violates the clinical responsibility to provide patients access to certain treatments [47]. Excluding participants with EOAD or including only EOAD participants might increase a study’s internal validity due to increased subject homogeneity; however, such could also consequently diminish a study’s external validity [48, 49]. Although preliminary trials on drugs and certain interventions are often done on more homogeneous populations as a result of relatively narrow selection criteria [50], participant recruitment for invasive neurosurgical procedures such as DBS could be extremely challenging, especially when highly stringent selection criteria are employed [2]. As such, recruiting an immensely homogeneous sample might not be possible in the context of preliminary DBS trials. Furthermore, trials including different populations for invasive procedures could provide better knowledge of different subpopulations that could be more responsive to treatments, provided that no subpopulation is significantly disadvantaged or harmed by the intervention in accordance with the ethics principle of Nonmaleficence [7, 51]. However, since initial results from the Phase II fornix DBS trials suggest that participants <65 could worsen from DBS [3], excluding them from subsequent fornix DBS trials, especially those with moderate AD, might be warranted until subsequent in-depth analyses have been made to ascertain if age is indeed the sole causative factor associated with the observed decline or if other variables such as genetic and cognitive status actually better explain the variable effects of treatment between different patient subgroups. This further emphasizes the importance of obtaining additional information on genetic status and biomarker information in participants so that those who are either likely to benefit or are likely to be harmed by DBS would be better and more precisely identified. Given that DBS for AD has not been approved yet by an established regulatory body (e.g., FDA) as a standard of care and its application is still in the context of clinical trials, denying access to it for certain patient subgroups could not be considered as denial of treatment.
Genetic vulnerability and informed consent
Since the corresponding clinical progression resulting from certain AD-associated mutations has already been recorded [23], making the correct adjustments such as allowable time period to withhold treatment and frequency of monitoring for patients with certain EOAD genetic subtypes would be much better facilitated if detailed genetic information is available. However, requiring genetic testing for autosomal dominant AD mutations in all or certain trial participants raises its own set of ethical issues, requires adjustments to the informed consent process, and entails additional procedures that have to be included in the trial (Fig. 1).
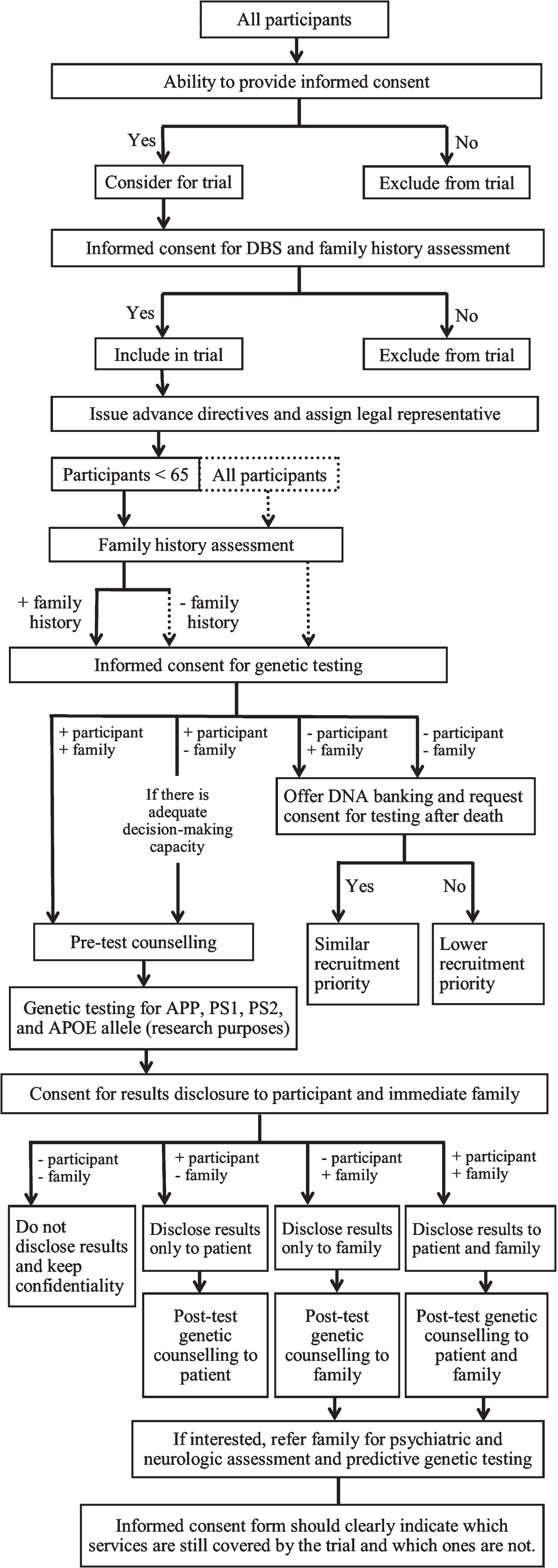
Decision tree for providing informed consent and for genetic testing in DBS trials including people with early-onset Alzheimer’s disease. Solid boxes, lines, and arrows indicate suggestions that must be minimally fulfilled. Dashed boxes, lines, and arrows indicate optimum suggestions.
First and foremost, even before having participants sign an informed consent form, it is important for all studies to assess their ability to consent using systematic or established measures of capacity [52, 53]. Participants who do not fully understand the risks of the invasive neurosurgical procedure or the associated uncertainty to benefit from the trial, given the limited data from preliminary studies, should be excluded.
Second, should genetic testing be required, it should be specified in the informed consent form that participants will consent to the DBS surgery and stimulation and all other pre- and post-clinical testing, including genotyping of APP, PSEN1, PSEN2, and/or APOE. The genotyping that will be performed should be clearly indicated. Both patients and immediate family members should be given options to decide whether they would like to have the results of the genetic tests disclosed or not. Since approved prevention methods [54] and treatment options for AD do not exist yet, there is no obligation for deliberate disclosure of genetic test results to participants and immediate family members [55]. Nonetheless, pre-disclosure genetic counselling using established guidelines [56, 57] should be provided to both patients and immediate family members to allow them to better understand the implications of results disclosure [58, 59], particularly on reproductive [60], insurance [61], and geriatric care planning and also on potentially being able to access certain clinical trials for those with autosomal dominant AD [62]. Should patients and/or immediate family members prefer to know results, further support in the form of post-disclosure genetic counselling sessions could be provided [57]. It should be emphasized that the risk of inheriting a mutation from a parent with autosomal dominant AD is 50%, and immediate family members who wish to undergo predictive genetic testing themselves should be referred to a genetic counsellor, neurologist, and psychologist/psychiatrist for further evaluation and support [56, 63]. Consequently, since payment for further testing and counselling sessions would raise financial concerns [64] for those conducting the trial and/or those participating in it, the extent of genetic counselling and compensation that will be provided should be clearly indicated in the informed consent form [51] to better allow family members to decide on whether they would still prefer to be informed of the results and understand potential limitations in the support that they would be receiving in the event of an unfavorable result. Finally, patients who do not consent to genetic testing should not be directly excluded from trials and should be offered alternative options such as family history assessment [65] for autosomal dominant EOAD risk estimation and DNA banking [56] for potential genetic testing after the patient’s death [66], with his or her consent. Those who do not consent to such alternatives might be given lower priority to participate in a trial.
Third, since a significant number of studies of DBS for AD [4, 32] required the consent of family members or caregivers, requiring genetic testing could raise potential issues when the patient but not the family members would consent to participation in the trial and consequentially, genetic testing [55]. Although at the start of the trial, participants with EOAD at a very early disease stage might still have adequate ability to consent, they might eventually need a caregiver when the disease rapidly progresses, and as such, caregivers’ opinions and support on a patient’s participation in a DBS trial [8] would be of increasing importance in later trial stages. This highlights a potential dilemma when there are conflicting opinions. The final decision on whether a participant should participate in a DBS trial and have a genetic test should then be made only after having a careful and collaborative discussion with the researchers, clinicians, family members, and the participant. If there are still conflicting opinions after the deliberation, we recommend that the decision of participants who have adequate decision-making capacity at the start of the trial be honored. Should the participant want to undergo DBS and genetic testing, legal representatives and advance research directives [7] should also be determined and set by the participant prior to DBS implantation. Given that several states and countries only allow consent on the subject’s behalf when the patient has a legal status of incompetence [67], legal representatives might have to make eventual decisions on whether to continue DBS stimulation; however, there might be a point prior to complete incompetence when the capacity to consent is uncertain and solely obtaining consent from caregivers would not be the best option from an ethical perspective [68]. In such instances, researchers could also seek assent from participants and exclude those expressing dissent [7], which may be indicated by signs or actions of frustration, unhappiness, discomfort, or passivity [69].
Disease progression and study design
Only two trials [4, 32] of DBS for AD have a case-control design, albeit with different durations in which stimulation was withheld from the control group. Withholding DBS for a long time from participants with EOAD might not be justifiable considering that DBS is an invasive procedure with potential unwanted side effects [34, 71], and people with EOAD might have a more aggressive disease course and higher mortality than LOAD [72]. It should also be taken into account that although the Phase I trial showed that patients who had milder cognitive symptoms at the time of DBS initiation have seemingly better metabolic and cognitive outcomes [1], the Phase II trial indicated in its subgroup analysis that participants <65 actually had worse cognitive scores after stimulation for one year, whereas the opposite was observed for those ≥65 [3]. Since results for each individual participant <65 were not presented, it is not yet known whether all of them experienced decline after DBS or if it is only a few with other characteristics such as lower cognitive profile or more advanced disease stage. In addition, it would be interesting to know if combining the results for the five participants <65 in the Phase I fornix DBS trial [1] with the results for those <65 in the Phase II trial [3] would still lead to the same observed differential effect of DBS for this subgroup. Given such uncertainty, it might be possible that some participants <65 could have also benefitted from the stimulation given that some participants <65 in the Phase I trial [1] had stabilized or improved cognitive scores following DBS. For those <65 who might benefit from stimulation and for the rest of participants who were in the control group, withholding stimulation for a year could potentially result to a loss of a significant number of time and treatment opportunities where their cognition could still be stabilized. Although patients will continue receiving medications for memory during the period without stimulation, limiting them from attaining any eventual potential benefit from the surgery and even potentially causing them harm should any untoward incident result from surgery or stimulation would be unfair. On the other hand, for those <65 wherein stimulation could be disadvantageous, having a shorter stimulation time instead of one year could have potentially allowed initial detection of the stimulation’s potential adverse effect, and appropriate actions could have been taken to prevent further harm in these patients.
The initial results of the Phase II trial also bring into question whether the study design has to be modified given that those <65 might be disadvantaged. It is important for Lozano et al. [3] to look at individual patients who deteriorated the most in the <65 stimulation group and see whether stimulation might need to be stopped for them instead of allowing them to continue to the one year open stimulation phase. In addition, those who are <65 who were originally assigned to the control group and have similar cognitive profiles as those who were most severely disadvantaged by DBS might not need to participate in the trial’s next phase and not receive any stimulation given that these participants might actually be harmed by it. Although these adjustments to the study design could have some effect on the study’s power if ever implemented, it is more important to protect participants’ welfare, especially if there is convincing evidence that their further participation could lead to avoidable harms.
In terms of patient monitoring, studies usually monitored performance in various cognitive and neuropsychiatric tasks a month, three months, six months, and a year after surgery. However, when participants with a potentially more rapid disease progression are included [23], more frequent monitoring (bimonthly or monthly) should be implemented. Given that those <65 who received stimulation for one year in the Phase II trial had worse outcomes than those who did not [3], much more frequent monitoring would have allowed the initial detection of this potential worsening and would have allowed more data to be obtained to determine the rate of disease progression and compare it with that prior to stimulation or with historical controls. Moreover, given that some people with AD-EOAD experience atypical symptoms [19] such as behavioral impairment, apraxia, and aphasia, stringent examination and careful neuropsychiatric monitoring before and post-implantation should be made to ensure that any neuropsychiatric or motor attributes would not be affected in a way that is detrimental to the patient.
Potential trial outcomes
All the trials that have been completed [1, 4] and the Phase II trial [32] that is ongoing assessed the efficacy of DBS using a cognitive test (MMSE, ADAS-Cog, CDR, FCSRT); however, several studies have also employed measurements of metabolism via PET [1, 32], brain activity through EEG [4], and changes in hippocampal volume through MRI [1, 32]. Some studies also assessed the participant’s quality of life [1, 32]. For the Phase I studies [3, 5], results on the quality of life have been inconsistent with an increase in some participants and a decrease in others. For the Phase II trial, Holroyd et al. [32] mentioned that they will be including the Quality of Life – Alzheimer Disease measure [73]; however, Lozano et al. [3] did not mention the result of this test in the report they have published [9]. All studies have also reported improved or preserved neurologic activity in certain brain areas; however, the translatability of these improvements to the trial participants’ quality of life and daily functioning has yet to be adequately proven.
Although in general, the final pathophysiology in EOAD and LOAD may be greatly similar, the initial disease progression and onset of EOAD and LOAD might have some differences that could lead to potentially variable outcomes during early disease stages. For instance, EOAD patients have more pronounced atrophy in neocortical areas as opposed to LOAD patients wherein atrophy is more severe in the hippocampus [74]. Moreover, EOFAD patients also present with apraxia, aphasia, or dysexecutive syndrome [19]. As such, additional modes of assessment should be performed in studies involving patients with EOAD to determine how DBS affects these cognitive domains and motor symptoms. It might also be possible that EOAD patients might have a different initial clinical outcome given that degeneration usually is not as prominent in the hippocampus. Depending on the target region for DBS, the extent of changes or duration of stabilization in cognitive scores could differ between EOAD and LOAD patients. Although the results of the Phase II trial might indicate that fornix DBS could potentially be disadvantageous for those <65 [3], such might not necessarily be the case for NBM stimulation. Given these, it is important to properly convey these potential sources of differential DBS response to EOAD patients, especially those with family history and mutation in APP, PSEN1, or PSEN2, so that they will be more informed when they consent to the procedure and also to increase the likelihood that they will monitor the effects of stimulation on these atypical symptoms once the trial has been initiated.
Interpretation and communication of study results
Results of preliminary studies are used to plan the next stages of clinical trials [75]; however, they should not be used to justify efficacy and safety [75, 76] in a clinical setting. It is important that trials should convey this in their discussion and conclusion to prevent creating false hype. In addition, they should also highlight limitations in their methodology that could have affected study results. For example, Kuhn et al. [4] mentioned that it proved impossible for them to precisely insert the electrode in their preselected target due to degenerative or pathological vascular alterations. Although they mentioned this as a limitation of their study, they should have reflected more on whether this limitation would then make precise targeting of a desired region in the NBM totally not feasible instead of just concluding that DBS of the NBM is “technically feasible”. In addition, they also mentioned that NBM DBS “apparently lacks significant stimulation-induced untoward effects”; however, they mentioned that one patient required lorazepam during the stimulation phase without fully describing why and at which exact points during the open stimulation phase was the drug prescribed. Finally, it is important to emphasize that the conclusion that DBS is “well tolerated” and that “four out of six patients were responders” should only be considered in the context of deciding whether to do a subsequent clinical trial in a proper and well-regulated research setting and not allowing for NBM DBS to be performed on anyone with AD in a clinical setting given that the study’s limited sample size is inadequate to capture a wide range of potential adverse events and derive any statistically valid conclusion on the efficacy of DBS.
Another important aspect in reports of trials is for authors to completely report the results of all statistical tests that they perform. In the results of the Phase II trial [3], the observed difference in results between those who received stimulation and those who did not is much more dramatic for those <65, whereas only slight improvements in cognitive function were observed for those who are ≥65. Although the authors mentioned the result of the statistical tests for the <65 group, they did not provide p values for the ≥65 group for readers to determine the significance of the observed decline between those who received and those who did not receive stimulation.
Recommendations for DBS clinical trials, especially those involving participants with EOAD, based on gaps in current and previous trials and case studies
Finally, it is important to consider that the 65 years cut-off point has no biological significance and is mainly based on employment and retirement age [15]. As such, it is crucial that further analysis for the Phase II trial [3] should be performed based on other factors such as disease stage, cognitive scores at the start of the trial, and/or extent of AD pathology based on biomarkers such as brain volume and levels of tau and/or Aβ in the cerebrospinal fluid [9, 46]. In addition, relating genetic data to treatment outcomes could potentially allow for better explanation of results obtained for those <65. It is possible that those who were made worse off by DBS have genetic mutations that result to a more aggressive disease course [16], and these individuals are also at a later disease stage at the trial onset. Reporting effects for participants having known mutations might require presentation of individual de-identified data to facilitate comparison of the rates of progression for individuals possessing mutations in known genes and accurately determine if DBS might have affected the rate of disease progression for these participants. Although introducing other variables in the analysis would add another level of complexity, they could facilitate improved understanding of the factors that affect DBS response, allowing better selection of suitable participants in future trials. Caution should be exercised in drawing conclusions though given that analysis based on subgroups usually lacks adequate power and may yield false-negative results, unless the initial trial power calculation significantly accounted for eventual subgroup analysis [77].
CONCLUDING REMARKS AND FUTURE CONSIDERATIONS
Studies that conducted DBS in patients with AD have not screened patients less than 65 years old for a family history of EOAD; mutations in APP, PSEN1, or PSEN2; or have APOE alleles that could affect age of disease onset and potentially, rate of progression [78]. As such, participants who have a familial or genetic AD that have a more aggressive disease course might have been disadvantaged by trials in terms of the employed study design and frequency of monitoring.
Although we believe that larger studies on DBS for EOAD should only be conducted after an extensive positive appraisal of the long-term results of the ongoing Phase II trial taking into account previous trials and relevant animal studies, we would like to propose certain precautionary recommendations for potential trials in the future that would include participants <65 (Table 2). First, in terms of patient selection, EOAD patients who have psychiatric disorders, suicidal ideations, and who are already at the dementia phase should be excluded in studies until results suggest that DBS might also be effective in later AD stages. Second, genetic screening of patients <65 years old should be included in trials; however, disclosure of results has to be discussed with patients and relatives. Third, appropriate adjustments on the length of exposure to trial arms, assessments performed, and frequency of monitoring should be made to accommodate differences in EOAD and LOAD should people <65 be included. Fourth, study results should be realistically conveyed and should be reported equally regardless of the direction of the effect. Researchers and the media should be careful not to hype up results of preliminary studies to ensure that EOAD patients volunteering to enroll in an experimental trial are fully informed and not just misled by overly positive depictions of DBS for AD [79]. Finally and most importantly, collaboration between basic researchers, neurologists, psychiatrists, neurosurgeons, genetic counsellors, ethicists, and other aged care personnel should be established to set a proper framework ensuring that patients with EOAD are appropriately prepared and informed, well-protected, unharmed, and are not deprived of potential therapeutic benefits in future clinical trials of DBS for AD.
Footnotes
ACKNOWLEDGMENTS
This work is supported by a Tasmania Graduate Research Scholarship awarded by the University of Tasmania to JNMV. MB was supported by the Federal Ministry of Education and Research of Germany (01GP1621A). FG is the recipient of an Australian Research Council Discovery Early Career Researcher Award (project number DE150101390). The Australian Research Council Centre of Excellence Scheme (Project Number CE 140100012) is gratefully acknowledged.