
Abstract
The nucleus basalis of Meynert (nbM) was first described at the end of the 19th century and named after its discoverer, Theodor Meynert. The nbM contains a large population of cholinergic neurons that project their axons to the entire cortical mantle, the olfactory tubercle, and the amygdala. It has been functionally associated with the control of attention and maintenance of arousal, both key functions for appropriate learning and memory formation. This structure is well-conserved across vertebrates, although its degree of organization varies between species. Since early in the investigation of its functional and pathological significance, its degeneration has been linked to various major neuropsychiatric disorders. For instance, Lewy bodies, a hallmark in the diagnosis of Parkinson’s disease, were originally described in the nbM. Since then, its involvement in other Lewy body and dementia-related disorders has been recognized. In the context of recent positive outcomes following nbM deep brain stimulation in subjects with dementia-associated disorders, we review the literature from an historical perspective focusing on how the nbM came into focus as a promising therapeutic option for patients with Alzheimer’s disease. Moreover, we will discuss what is needed to further develop and widely implement this approach as well as examine novel medical indications for which nbM deep brain stimulation may prove beneficial.
FROM DISCOVERY TO FUNCTIONAL CHARACTERIZATION
The goal of this review is two-fold. On the one hand, it gives an historical overview of the nucleus basalis of Meynert (nbM) description and function and how it came to be considered for deep brain stimulation (DBS) in the context of cognitive disorders, especially dementia. On the other hand, the goal of this review is to restate the importance of pursuing nbM DBS in patients suffering from Alzheimer’s disease (AD), discussing its improvement over time, and how it can be further refined. We follow the key findings in a chronological order to better highlight the progress made in nbM DBS and its future direction.
The basal forebrain is comprised of a complex ensemble of heterogeneous structures lacking layered organization, hindering the classification of its structures. Theodor Meynert described a group of cells located within the substantia innominata and coined this structure the ganglion of the ansa peduncularis [1]. Albert von Kölliker provided the first detailed description of this structure, noting a group of highly-pigmented cells, 20–30 mm in size and named it Meynert’s ganglion basale [2]. The intricacy of this area has led to multiple names for this cluster of cells, resulting in substantial inconsistencies across studies [3–5]. Ultimately, in 1942, the term nucleus basalis of Meynert was established and is now widely used [6] (see Fig. 1).

T2 coronal MRI image showing the electrodes leads (white dots) with the nucleus basalis of Meynert boundaries of a patient suffering from Alzheimer’s disease. Put. putamen; Gpe, external Globus pallidus; Ch4, Nucleus basalis.
The nucleus basalis is well formed and extensive in certain species such as dolphins (Delphinus delphis), but sparsely distributed and less organized in other mammals such as rats (Rattus norvegicus), in which it is referred to as nucleus basalis magnocellularis [7]. In humans, the nbM is a structure extending from the olfactory tubercle to the level of the hippocampal uncus. The anterior part of the nbM reaches the horizontal limb of the nucleus of the diagonal band of Broca ventrally, the ventral globus pallidus medially, and the lateral extension of the anterior commissure laterally. The nbM posterior portion reaches the ansa lenticularis dorsally, the putamen laterally, the posterior tip of the amygdala ventrally, and the optic tract medially [8]. In terms of size, the nbM has been measured to span approximately 13-14 mm in the anteroposterior axis with its widest mediolateral extent within the substantia innominata covering 16–18 mm [9]. It contains about 200,000 neurons per hemisphere, with the highest neuron density under the anterior commissure [10]. Based on the distribution of acetylcholinesterase (AChE) and choline acetyltransferase (ChAT) staining, the nbM can be subdivided into neuronal clusters that form anteromedial (am), anterolateral (al), antero-intermediate (ai), intermedio-dorsal (id), intermedio-ventral (iv), and posterior sectors (p) [11].
There are no intrinsic cholinergic neurons in the human cerebral cortex, amygdala, or hippocampus. In investigated cortical areas, ChAT-positive and nerve growth factor (NGF) receptor-positive axons show similar patterns of distribution and density, suggesting the cholinergic innervation almost exclusively stems from Ch1–4 [12]. The Ch1–4 nomenclature was introduced to designate ChAT-containing neurons (i.e., cholinergic neurons) belonging to four continuous groups of cells of the basal forebrain [13]. The Ch1 and Ch2 sectors are contained within the medial septal nucleus and the vertical limb nucleus of the diagonal band, respectively. The Ch3 sector is contained mostly within the lateral portion of the horizontal limb nucleus of the diagonal band. Finally, the Ch4 sector is comprised of cholinergic neurons in the nucleus basalis and parts of the diagonal band nuclei. Cholinergic Ch4 neurons are mixed with a heterogeneous population of non-cholinergic neurons [14]. Therefore, the terms Ch4 and nbM are not synonymous: nbM is used to designate the entire neural population of the nucleus, whereas Ch4 refers to its contingent of cholinergic neurons. Nearly 90% of nucleus basalis neurons are cholinergic.
Cholinergic neurons of the nbM express estrogen receptors (ERα and ERβ), glutamate receptors and calbindin, as well as m2 muscarinic acetylcholine receptors [15, 16]. The 10% of Admixed non-cholinergic interneurons of the nbM include galaninergic neurons, which inhibit cholinergic neurons, and nicotinamide adenine dinucleotide phosphate-diaphorase (NADPH-d) containing neurons, implicated in NO synthesis [17, 18]. Furthermore γ-aminobutyric acid (GABA)-ergic neurons are distributed throughout the basal forebrain [19].
The detailed topography of Ch4 projections remains largely unknown in humans. In the rhesus monkey, individual cortical areas receive their major cholinergic input from different sectors of the Ch4 complex. For example, Ch4am provides the major source of cholinergic input to medial cortical areas, including the cingulate gyrus; Ch4al projects to the frontoparietal cortex, opercular regions, and the amygdaloid nuclei; Ch4i projects to the laterodorsal frontoparietal, peristriate, and midtemporal regions; and Ch4p projects to the superior temporal and temporopolar areas [13]. All cortical areas receive Ch4 projections, but not all cortical areas project back to Ch4. In the rhesus monkey, tritiated amino acid injections in various cerebra showed anterograde transport to Ch4 from the piriform cortex, orbitofrontal cortex, frontopolar cortex, anterior insula, temporal pole, entorhinal cortex, and potentially the anterior cingulate as well. Ch4 also receives inputs from the hypothalamus and amygdala [20]. The selectivity of cortical projections to the nucleus basalis remains to be fully described in the human brain.
Recent breakthrough research efforts studying this brain region have resulted in multiple lines of evidence supporting that a decline in the connectivity number, size, or function of cholinergic neurons in the nbM may be linked to cognitive impairments observed in AD, other forms of dementia, and even healthy aging [10, 21–24].
Postmortem studies have shown that loss of cholinergic neurons is associated with neurofibrillary tangle (NFT) neurons in the nbM [25]. Furthermore, a correlation between cholinergic abnormalities and amyloid-β (Aβ) pathology have been long established [26]. Human studies have shown that cholinergic abnormalities occur as early as asymptomatic or at prodromal stages of AD and basal forebrain atrophy could be reduced by AChE inhibitors [27]. The cholinergic degeneration associated with basal forebrain atrophy in the nbM is mainly pre- rather than postsynaptic [28]. This observed decline of cholinergic neurons in the basal forebrain is driven by a decline in release of acetylcholine (ACh) and decreased activity of ChAT and AChE [24]. Interestingly, AChE activity levels are increased around NFT neurons, likely driven by Aβ42 and mediated via, among others, oxidative stress [29]. NGF is currently viewed as the most relevant trophic factor for cholinergic neurons of the basal forebrain and is downregulated in the basal forebrain nuclei in AD, driven by decreased expression and immunoreactivity of the TrkA high-affinity receptor. Neurons in the nbM require NGF, TrkA, and pan-neurotrophin receptor P75 for maintenance and survival [30]. TrkA expression is downregulated in patients with mild cognitive impairment and mild AD, indicating that the necessary neurotrophic support for nbM neurons to maintain function is lacking [31]. Tiernan and colleagues show downregulation of transcripts encoding TrkA, TrkB, and TrkC in individual nbM neurons, with presence of the pre-tau marker pS422. This suggests that nbM neuron survival is compromised before mature NFT formation is present. The authors point out that they cannot establish causality, but propose phosphorylation of tau at S422 and inhibition of retrograde transport may impair positive feedback from NGF, leading to diminished TrkA expression in nbM neurons [32]. Recently epigenetics has gained more attention regarding its role in cholinergic nbM neurons in AD. Mahady et al. show that cortical histone deacetylase (HDAC) dysregulation contributes to cholinergic nbM dysfunction. Both histone acetylation and deacetylation are involved in regulating gene expression of ChAT, indicated by decreased expression of basal forebrain HDAC2 and ChAT protein with progressing AD [33].
Between the two major hallmarks of AD, compromised of Aβ42 and NFT, there are multiple possible modes of interaction between the two hallmarks. Firstly, Aβ drives tau toxicity, by causing hyperphosphorylation of tau. Secondly, tau mediates Aβ toxicity. Thirdly, there is a synergistic interaction where Aβ and tau interact on different sites of the same organelle [34]. The nbM is affected early in the progression of AD, presenting more NFTs than other brain regions of the brain in early or pre-symptomatic stages of AD, especially in Ch4p [35].
The nbM is part of an uninterrupted band of non-isocortical basotemporal areas (e.g., entorhinal region, hippocampal formation, amygdala, and piriform cortex) where the tauopathy and neurofibrillary degeneration of AD pathology originate, and from which they seem to spread centrifugally toward other parts of the cerebral cortex [36]. The nbM is considered to be the telencephalic extension of the brain stem reticular formation and, functionally, it has been linked to attention, arousal, and memory due to its hippocampal input [37].
FIRST CLINICAL REPORT
Whitehouse and colleagues showed evidence for selective loss of cholinergic neurons in the basal forebrain after autopsy of a patient suffering from AD [38]. In 1985, Ian Turnbull and colleagues devised a novel approach to alleviate AD-related senile dementia targeting the nbM [39]. They implanted a flexible electrode targeting the left nbM (50 Hz; 3 V; 210 ms pulse width; duty cycle 15 s/12 min ON/OFF) in a patient suffering from AD to increase cortical glucose metabolic activity and improve the clinical symptoms of AD [40]. Following 8 months of nbM stimulation, the authors reported no immediate clinical effect, but a decreased cognitive deterioration compared to what was expected based on the natural course of the disease. Additionally, the authors reported less of a decrease in glucose utilization as measured by PET scan one-month post-surgery on the ipsilateral side of the stimulation as compared to the contralateral side, using PET scan measurements from pre-implantation state as baseline. The results of this clinical report are difficult to interpret as the severity of the symptoms is not clear, the reported parameters of stimulation are inconsistent, and there is no mention of changes in quality of life. Nevertheless, this was a pivotal study in the field of DBS. First, it was one of the first attempts to use DBS on neuropsychiatric disorders, a technique at the time considered almost exclusively for movement disorders [41]. Second, the authors used glucose metabolism as an objective measurement to evaluate the effect of their treatment. Thirdly, it was the first study, to our knowledge, to investigate the potential of low frequency DBS.
The authors report a second CT scan showed the lead was within a millimeter of the intended target (nbM) during implantation, but that during internalization of the stimulation system the electrode was retracted. A lateral radiograph of the skull revealed that the first site of stimulation was the nucleus accumbens. The electrode was subsequently repositioned on target. Interestingly, it provides the first evidence that electric stimulation at 50 Hz of the nucleus accumbens is safe, as no behavioral effect were noted. Nowadays, this structure is one of the main targets for DBS in patients with obsessive-compulsive disorder [42]. The authors did not explain their choices of stimulation pattern, which induces a very small amount of current, compared to more recent studies, and may not have stimulated cholinergic release in a clinically quantifiable way. On the other hand, early applications of DBS therapy in movement disorders produced a dramatic and immediate clinical effect. Following this seminal clinical case report, research on the nbM intensified due to its central involvement in the cholinergic hypothesis of geriatric memory dysfunction [23]. In the next two decades following the first case report, research on the nbM focused on pre-clinical work.
PRE-CLINICAL WORK FROM 1983 TO 2009
The nbM can influence cholinergic transmission throughout the cerebral cortex, affecting the neural activity of some of the components of the limbic system. In Fischer rats, performance deficits induced by nbM lesions via ibotenic acid were initially interpreted as being the result of impairments in learning and memory abilities [43–45]. However, single-unit recordings in non-human primates revealed that nbM neurons are selectively sensitive to stimuli with motivational relevance, such as visual stimulus associated with reward delivery [46, 47]. Since then, nbM lesion studies became more specific and accurate in their targeting [48, 49] and the role of the nbM in cognitive function became more precise. Several studies support its involvement in specific aspects of attention in rodents [50–54] and monkeys [55, 56]. A growing body of evidence suggests attentional deficits to play a major role in the cognitive deficits associated with lesions to the nbM and not directly to learning and memory itself. Attention can be described as the focused neuronal activation of a neuronal network that is relevant to a specific cognitive task [57]. The reported deficits from ibotenic acid lesion studies on learning and memory could not be replicated using more potent specific cholinergic excitotoxins such as AMPA. The observed deficits are likely to be attributed to the disruptions of cortico-striatal outputs passing through the dorsal and ventral globus pallidus [51]. A selective role of the basal forebrain via cholinergic transmission in the modulation of attention is suggested [58]. Basal forebrain lesion studies have shown different forms of attention to be disrupted [59]. Rats with neurotoxic lesions to the nbM perform worse on a multiple-choice reaction time task. Furthermore, the effect could be manipulated based on how long the relevant stimulus was shown [51]. A study by Chiba and colleagues showed cue target detection to be altered by selectively removing cholinergic neurons of the basal forebrain [60]. A more recent line of research has shown that response to novelty and surprise-timing, both requiring sudden attention, is regulated by the basal forebrain, namely via phasic bursting and tonic ramping [61]. A review by Blokland and colleagues on the role of the cholinergic system and attention, pooled data from animal and human studies and proposes that the role of ACh on cognition changes, depending on the brain area [59]. This expanding body of research into attention further strengthens the pivotal role of the basal forebrain for cognitive functioning. It is important to note, that while the nbM has gained more consideration in its role in attentional deficits, early research on the involvement of ACh on attention focused on the role of the reticular ascending cholinergic system [62]. Other brain regions, such as the prefrontal cortex, the parietal cortex, hippocampus, and amygdala are involved in modulating attention as well. For an overview of involved brain structures in modulating attention, see Blokland et al. [59].
In order to identify which inputs modulate the activity of cholinergic neurons and lead to changes in ACh output from the cerebral cortex, Fiorella Casamenti and colleagues applied electrical current directly to the nbM in Wistar rats [63]. They measured an average of 40% increase in ACh release in the parietal cortex when stimulating (30 Hz 0.1 mA 0.25 ms 1 s per 3 s). Furthermore, Biesold and colleagues demonstrated for the first time that stimulation of the nbM could modulate cortical blood flow [64]. In this study, the recording probe of a laser Doppler flowmeter monitored the parietal cortex blood flow during unilateral nbM stimulation (50 Hz, 10–1000 μA, 0.5 ms 10–20 s) in Wistar rats. Several locations of stimulation within and outside the nbM were probed. No response was observed when the stimulus was lower than 50 μA, with the most robust effect, a 150% increase in cortical blood flow, was reported following a 200 μA stimulus within the edge of the nbM. Using several cholinergic blocking agents, permeable or not to the blood-brain barrier, it was determined that the induced vasodilative response was likely due to an increase in ACh secreted from cholinergic nbM-projecting neurons. Later that year, the same group also established that unilateral stimulation of the nbM induced an ACh release in the parietal lobe [65]. The ACh release was proportional to the intensity of the stimulus and its frequency, up to 20 Hz, at which point it reached a maximum plateau. The vasodilative effect of nbM stimulation was later confirmed and extended to frontal and occipital cortices, using different measurement methods while extending the stimulus duration to 60 s [66, 67]. Cholinergic fibers originating in the nbM do not contribute to neural vasodilatation of the pial arteries, although they induce neural vasodilation of potentially parenchymal blood vessels, resulting in increased cerebral blood flow (CBF) in the parietal cortex when stimulated at 50 Hz [68]. The involvement of parenchymal blood vessels was confirmed in another study of the same group. Moreover, administration of a nitric oxide (NO) synthase inhibitor dampens the vasodilative effect of low frequency nbM stimulation, thereby suggesting that NO is a key player of the induced response [69].
Douglas Rasmusson and colleagues contested, however, the fact that low frequency induced the strongest cortical ACh release, reporting an increase twice as large in magnitude when stimulating at 100 Hz, as compared to lower (10 and 50 Hz) or higher (200 Hz frequencies) [70]. These different findings can be explained by different methodological approaches. While Kurosawa and colleagues show that ACh release to be the strongest when stimulating the nbM at 20 Hz, rats were stimulated continuously, which could have led to ACh depletion with increasing frequency [65]. Rasmusson and colleagues stimulated rats for 10 min in 2 stimulation periods, showing the highest increase of cortical extracellular ACh in the parietal cortex at 100 Hz [70]. Comparing these two studies highlight some of the intricacies when exploring DBS stimulation parameters.
The CBF response to nbM stimulation is age-dependent. In a study performed in awake rodents, nbM stimulation (100 Hz; 50 μA; 0.5 ms pulse width, in continuous 1 s on/1 s off trains) induced a lower vasodilative response in older (22–28 months old) rats compared to younger rats (2–4 months old), both in the parietal and frontal cortices [71]. This study also reported that the vascular changes are strongest in the frontal cortex and are associated with low metabolic changes (glucose level), unlike in subcortical structures like the thalamus where metabolic changes are strong. Another study suggests that this age-dependent effects of nbM stimulation is further dampened in elder rats (32–42 months old) when stimulating (50 Hz; 20–200 μA for 90 s) [68]. The CBF lesser response to nbM stimulation was associated (to a lesser extent) with a less pronounced ACh release in the parietal cortex. The response to muscarinic ACh receptor agonists is unchanged with age, suggesting this lessened effect on the CBF may be mediated by nicotinic receptors, known to be involved in both the vasodilated cortical process and decline over time [72].
As nearly 90% of neurons in the nbM are cholinergic, a substantial body of research has investigated the role of cholinergic neuron receptors. Whitehouse and colleagues show a reduction of nicotinic acetylcholine receptors (nAChR) in patient brains of AD [73, 74]. Specifically, α4β2 nAChRs are affected by AD pathology, and have been implicated with upregulation of plaques and NFTs [75]. Moreover, α4β2 nAChRs can be upregulated by chronic nicotine treatment in rats [76], while protein content of α3, α4, and α7 of nAChRs are reduced [77]. Decrease of α7-nAChR is known to impair memory, and increased exposure to nicotine has been shown to improve cognitive function in patients suffering from AD [78]. Furthermore, activation of α7-nAChR function has been shown to alleviate Aβ toxicity, by inhibiting amyloid plaque formation [79].
Low frequency stimulation of the nbM produces an increase in extracellular levels of NGF in the cortex. The NGF levels, which are stable at rest, increase in the ipsilateral cortex from 200–500 min after the end of focal electrical stimulation to the unilateral nbM for 100 min [80]. This effect is sensitive to nicotinic ACh receptor antagonists that cross the blood-brain barrier. Incidentally this effect is absent in 29–31-month-old animals [81].
The nbM is shown to be indirectly involved in memory formation, as discussed above, based on lesion studies. This role is also supported by electrophysiological findings. Attention to biologically relevant stimuli was associated with an increase in the activity of basal forebrain neurons in primates and rats [82, 83]. Single units within the nbM and the nucleus of the diagonal band of Broca in both Wistar rats and Rhesus monkeys were selectively responsive to visual or auditory stimuli that were associated with the expectation of food reward [47, 84]. Firing rate of nbM single units increases when an object associated with a reward is presented and decreases strongly when the object is withdrawn [47]. The recorded nbM cells are assumed to be cholinergic neurons because they could be antidromically driven by cortical stimulation. Putative cholinergic neurons could increase the efficiency of cortical processing when decisions are being made regarding the appropriate behavioral responses to specific salient stimuli [82]. The corticopetal nbM system is involved in the control of shifting attention to potentially relevant, brief sensory stimuli that predict a biologically relevant event. To provide a direct link between activation of the nbM and memory, Dewey McLin and colleagues designed an experimental schedule aimed at inducing associative memory by pairing a tone with nbM stimulation in the absence of a reinforcer [85]. Adult male Sprague-Dawley rats were trained either with paired tone (6 kHz) associated with nbM stimulation or only with the paired stimuli, to control non-associative factors. They controlled the specificity to the newly formed associative memory to the 6 kHz tone by later measuring physiological responses of trained rats to other tones of a different frequency. Heart and respiration rates were used as behavioral indicators for the newly formed memory, and EEG was recorded with a specific focus on the gamma band power (30–58 Hz,). Stimulated subjects showed frequency generalization gradients, with a peak of 6 kHz, for both cardiac and respiratory measures, and the gamma band relative power level peaked for 6 kHz tones. By contrast, unstimulated subjects did not exhibit such generalization gradients or gamma band power sensitivity to a specific tone frequency. Unfortunately, the authors reported little on the stimulation pattern. Their description suggests that a constant current was applied for 2 to 5 s at increasing amplitudes (50–100 μA), until an EEG activation was observed (i.e., a shift from higher-voltage slower waves to lower-voltage faster waves).
Consistent with the studies on salience and memory formation in an aversive context, nbM stimulation promotes avoidance behavioral acquisition, but does not alter retention performance when the stimulation occurs after the avoidance training or before the retention testing. In these studies, adult male Sprague-Dawley rats were stimulated unilaterally in the nbM for 20–45 min (1 Hz; 60–100 μA; 0.5 ms pulse width) immediately before or after acquisition training, or immediately before retention sessions (24 h, 48 h, 11 d after acquisition training) [86, 87]. The same group also tested the ability of nbM stimulation to promote memory formation using a relational odor-association task [88]. Rats were stimulated as reported in their earlier work before the social training. They were also tested on their ability to remember trained food preference, either immediately or following a 24-h delay. nbM stimulation improved retention regardless of delay and correlated with an increase of c-Fos activity in the medial prefrontal and orbitofrontal cortex, as well as in some hippocampal sub regions. No such changes were observed in the basolateral amygdala, dorsal dentate gyrus, ventral Cornu amonis, or ventral subiculum.
Altogether, preclinical research in the early 2000 s suggested that modulating the nbM activity with stimulation at low frequency may have a therapeutic effect in neurological conditions such as dementia, albeit with an altered pattern of stimulation than the original clinical attempt by Turnbull in 1985 [39].
CLINICAL TRIALS FROM 2008-2015
In 2008, Sturm and colleagues implanted single bilateral electrodes into the nbM, in addition to electrodes placed in the subthalamic nucleus (STN), in a 71-year-old man with slowly progressive Parkinson-dementia syndrome [89]. The tips of the nbM electrodes were directed into the laterodorsal portion of the intermediate sector of the nbM, in the area in which the anterior commissure and the optic tract cross in a horizontal section. The patient displayed cognitive decline over the last 2 years that intensified over the last 6 months. The cognitive decline was mainly characterized by apathy, rigid thinking, poor short-term memory, slowing of thought processes, an impaired capacity to use acquired knowledge, and a lack of concentration. The bilateral subthalamic stimulation at high frequency was aimed at reducing the motor symptoms of Parkinson’s disease (PD), while the bilateral low frequency stimulation (20 Hz) of the nbM was aimed at reducing the severity of his neurocognitive symptoms. The neurological battery tests were conducted before surgery and after treatment with DBS in four double-blind phases (alternating combined STN-nbM stimulation with sole STN stimulation). The overall follow-up lasted up to 29 weeks post-implantation. At the 13-week follow-up after nbM stimulation, the authors reported a stabilization of the cognitive decline with clear improvements in various aspects of cognitive functioning such as attention, concentration, alertness, drive, and spontaneity. Improvements induced by nbM stimulation deteriorated in the intermittent nbM-off 1-week phase. The authors reported that improvement of personality features and social communication were more consistent than selective memory enhancement, as memory improved but remained deficient.
This clinical report was the first to demonstrate success in improving cognitive symptoms during nbM stimulation. Freund and colleagues stimulated bilaterally and at lower frequency (20 Hz; 1 V; 120 μs pulse width). Neurons within the nbM typically have a low-frequency firing rate, and low-frequency stimulation is believed to activate cell bodies [90, 91]. Moreover, nbM stimulation seemed to induce a generalized effect on cognitive performance and personality. Building on this success, low frequency stimulation of the nbM was investigated in a phase I trial for alleviating cognitive symptoms in AD patients (ClinicalTrials.gov Identifiers: NCT01094145).
A more recent randomized double-blind crossover clinical trial by Gratwicke and colleagues implanted bilateral electrodes into the nbM for 6 patients suffering from Parkinson’s disease dementia (PDD). Patients were stimulated at low frequency (20 Hz) for 6 weeks or sham-stimulated for 6 weeks. After 6 weeks, conditions were switched. No consistent improvement was observed in primary cognitive outcomes, however an improvement on the neuropsychiatric inventory was observed during stimulation as opposed to sham stimulation [36]. All critical scientific works necessary to justify the first clinical trial are presented in Fig. 2.
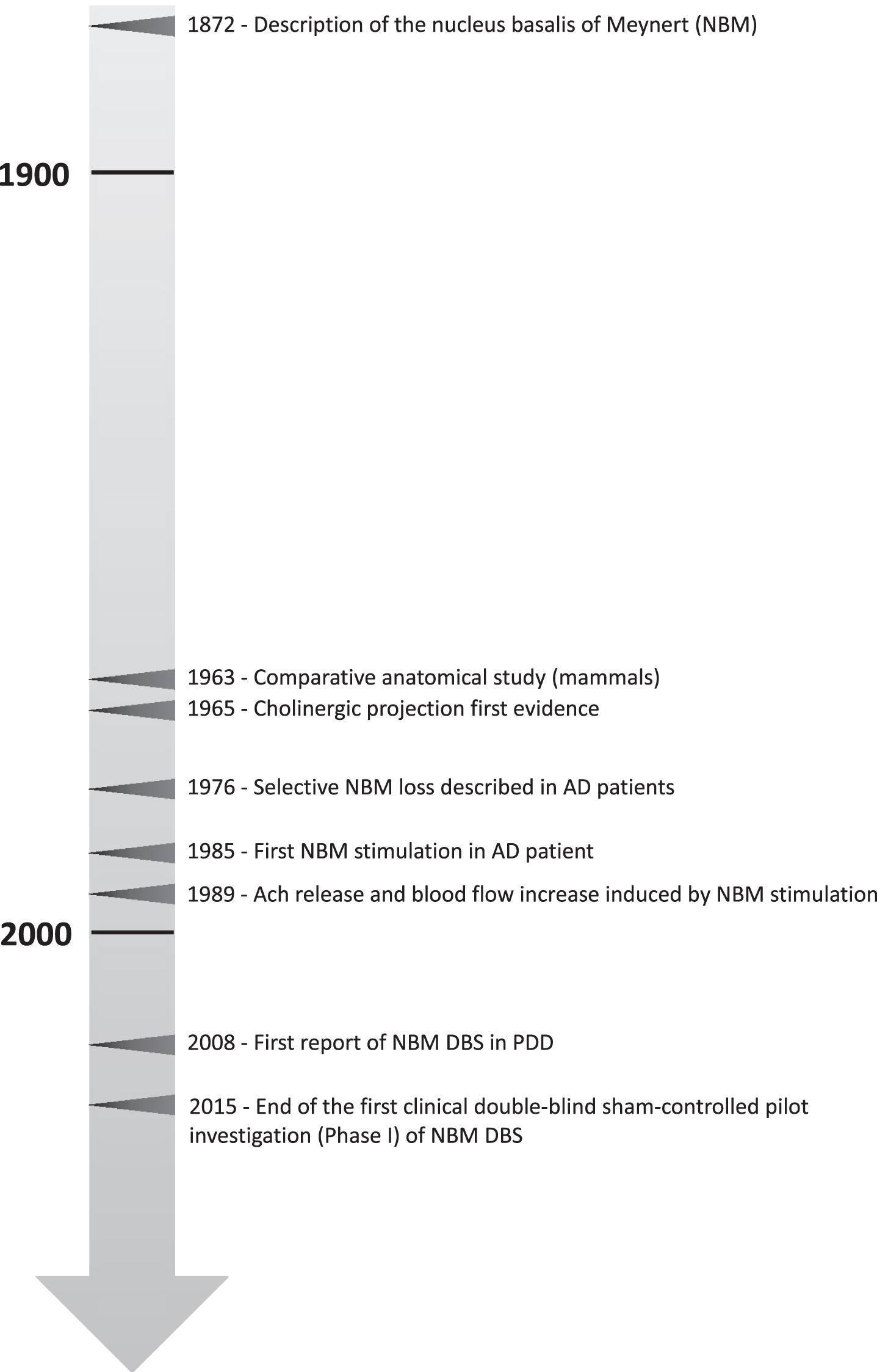
Timeline of selected key scientific work that contributed to establish deep brain stimulation of the nucleus basalis of Meynert (NBM) as therapeutic strategy in the context of dementia disorders. AD, Alzheimer’s disease; Ach, acetylcholine; DBS, deep brain stimulation; PDD, Parkinson’s disease dementia.
The results of the first phase I trial double-blind sham-controlled pilot testing the efficacy of DBS of the nbM for the treatment of cognitive deficits in mild to moderate AD are now available [92–94]. Depending on the patient’s unique anatomy and their specific pathological vascular alteration (or other deteriorations associated with disease progression), the final placements of the stimulating electrodes lay within the Ch4id, Ch4ip, or Ch4p. These studies suggest that nbM stimulation tends to halt the progression of the disease at the time of implantation. Specifically, Alzheimer’s Disease Assessment Scale cognitive subscale scores, a cognitive impairment assessment scale designed for AD patients, worsened by an average of three points after one year of stimulation, which is considered non-clinically relevant. This stabilization of cognition in three patients was associated with an increase in cerebral glucose consumption [94]. In line with this conclusion, stimulation of the nbM had no significant effect on the nutritional status of AD patients [95]. In two other, younger male patients with moderate AD, nbM stimulation at low frequency had a favorable impact on disease progression and cognitive functions. This was especially apparent in one patient, who showed cognitive improvements after 26 months of stimulation. The authors speculate that nbM stimulation modulates cholinergic input and possibly via M1 receptor stimulation and even the production of neurotoxic Aβ plaques, thereby slowing down the progress of the disease [92].
Stimulation of the nbM can modulate Ch4 neurons ACh release or trigger cholinergic-induced increases in blood flow and NGF secretion in the cortex, at low frequency, and these mechanisms can be co-occurring as discussed before. ACh binds to two major receptor types, ion channel nicotinic receptors and G protein coupled muscarinic receptors. At the same time, ACh is acting on cells, both neurons and glia that are adjacent to blood vessels and that contain NO synthase in rats [96]. Cholinesterase inhibitors in humans induce changes in blood flow of 10–15% [97]. CBF is typically reduced in AD, and reductions in blood flow likely contribute to the cognitive impairments. As the blood flow increases caused by basal forebrain stimulation are entirely parenchymal, the neuropil will be perfused with greater metabolic nutrient supply [98]. The hypothesis that blood flow causes a cognitive change is also bolstered by the fact that NO synthase inhibitors have been shown to have a deleterious effect on performance in a working memory task in non-human primates [99]. NO synthase inhibitors will impair the cholinergic modulation of blood flow modulation without directly affecting the cholinergic modulation of neurons. Nicotinic receptors mediate the vascular effect of nbM stimulation and their concentration decreases with age naturally, even more rapidly in the case of AD patients [72, 101]. Thus, the vasodilatation effect of nbM stimulation in older patients is likely to be less prominent in early onset AD patients. This can partially explain the difference in treatment efficacy among patients observed in the phase I trial. Additionally, it suggests that ACh-induced increased cortical blood flow is an important component for optimal treatment effectiveness. In turn, this specific effect induced by nbM stimulation would predict that younger patients are likely to be better responders to nbM DBS.
As AD progresses, nearly all neurons in the nbM become invaded by NFTs, eventually forming ghost tangles, remnants of dead neurons. At this stage, cortical cholinergic axons are severely depleted. The nbM is not a site of particularly prominent amyloid deposition, and there is no correlation between plaque density and severity of cholinergic axonal loss in cortical areas [25]. The cholinergic lesion in AD is a marker of the tauopathy present in AD patients. The cholinergic denervation in AD is usually most extensive in the temporal lobe, including the hippocampus, the entorhinal cortex, and the lateral and accessory basal nuclei of the amygdala, but much less conspicuous in primary sensorimotor cortices [102]. Although the nbM is small, it is the source of dense projections to all cortical areas. Its vulnerability to neurofibrillary degeneration creates a setting in which the earliest stages of AD pathology can impact the physiological integrity of the entire cerebral cortex and might provide the best window of intervention for nbM DBS, as suggested by the progression of the tauopathy in case of AD patients [103]. The putative effects of the nbM stimulation are shown in Fig. 3.

Schematic representation of the putative effect of deep brain stimulation (DBS) of the nucleus basalis of Meynert. Intra- and extra-synaptic effects of acetylcholine (ACh) are shown. Top left shows potential effect of DBS by altering cholinergic neurotransmission. Bottom left shows vasodilative effects of DBS via cholinergic activation. Right side shows nbM projection sites after DBS. NGF, nerve growth factor.
WHAT’S NEXT?
As nbM DBS has reached clinical trials to improve patient care, pre-clinical work in recent years has explored more on the potential of nbM DBS and how to refine its potential use for patients suffering from AD. Blake and colleagues have published several articles on stimulating the nbM with intermittent DBS in rhesus monkeys to showcase the superior cognitive benefits in terms of working memory and sustained attention when compared to continuous stimulation [104]. These findings highlight potential refinements to nbM DBS to further improve patient care. However, there is more research needed to validate the efficacy of this new stimulation pattern in an animal model for dementia [105].
Animal research focusing on fornix stimulation has provided more supporting evidence for the efficacy of DBS in AD [106–109]. To date, we only know of a single study directly addressing the question of DBS of the nbM on memory function in an animal model of dementia [110]. In this study, the authors report an improvement in spatial memory after nbM DBS in a pharmacological rat model for dementia. Stimulation was associated with changes in glutamatergic and GABAergic metabolism in the medial prefrontal cortex and the hippocampus, as well as changes in AChE activity in the same structures. However, there are several important limitations to this study. The model used in this study is the denervation of basal forebrain cholinergic neurons with 192-IgG saporin. The interpretation of this model is limited due to the nonspecific cell loss within the cerebellum and its potential repercussion on swimming behavior as a side effect [96]. The authors stimulated unilaterally at a high frequency (120 Hz). Most accumulated knowledge to date on the effect of nbM stimulation originates from studies using low frequency stimulation. The results are promising; however, they are difficult to reconcile with the rest of the current literature. More preclinical research is needed to provide an empirical basis to pursue clinical trials. One limiting factor is the ability to adequately and reliably mimic certain aspects of dementia. In that regard, rodent studies, which test the potential of DBS for cognitive enhancement, often use animal models for AD that are episodic and non-progressive [109, 111–117]. In contrast, several promising transgenic rat models for AD have recently emerged [118], in particular the TgF344-AD rat line that produces a progressive neurodegeneration analogous to AD patients [119]. This model manifests age-dependent cerebral amyloidosis that precedes tauopathy, gliosis, and apoptotic loss of neurons in the cerebral cortex and hippocampus, and cognitive disturbance. TheTgF344-AD rat fulfills a critical need for a next-generation animal model to enable basic and translational AD research. Preclinical research has relevant advantages compared to clinical trials such as generating new stimulation patterns that may yield better results with fewer side effects or usage of battery [104].
Additionally, preclinical work allows for significantly bigger cohorts to be tested, while allowing for more invasive data collection (e.g., multiple site of recording with a greater number of deep recording electrodes). Finally, animal models enable immediate postmortem quantification of various biomarkers relevant for AD through biochemical and histological techniques.
Neuronal loss in the nbM is established in dementing disorders. However, its pathological significance was first recognized in PD patients by Friedrich Lewy where severe neuronal degeneration and intraneuronal tangles were observed (1913). These intraneuronal inclusions were later given the name Lewy bodies (LB) and described first in the nbM. Moreover, the nbM has been involved in many other neuropsychiatric disorders including schizophrenia [120], Pick’s disease [121], Creutzfeldt–Jakob disease [122], Down’s syndrome [123], and Rett Syndrome [124–126]. The most documented consequences of the degeneration of the nbM are reported in the cases of PD and AD patients [127], although the cause of neuronal loss in the nbM might differ depending on the condition [128]. Moreover, nbM neuronal loss in PD has been reported to be more extensive, but without tau NFTs unlike in the nbM of AD patients [129]. Tau NFTs are more closely associated with cognitive decline [130] than Aβ plaques, the other main neuropathological hallmark of AD. LB- and AD-type pathologies co-exist in neuronal tissue of the brains of PDD and dementia with Lewy bodies (DLB) [131]. Moreover, there may be synergistic relationships between the two types of pathologies in the development of dementia [132]. Other disorders involving nbM malfunction may therefore benefit from nbM DBS, such as DLB [133].
From the clinical perspective, results of the first phase I trial are promising. For a review regarding clinical trials on DBS in dementia, see Xu and Ponce [134]. Currently, there are six ongoing clinical trials exploring the potential of nbM DBS for PDD and DLB (ClinicalTrials.gov Identifiers: NCT02263937, NCT02589925, NCT02763397, NCT01340001, NCT02924194, and NCT02369003). Regarding clinical trials, ethical issues must be considered. It is important to note, that while nbM DBS is a safe procedure, there are possible complications and side effects, including hemorrhage (1.3–4%), infection (2.8–6.1%), electrode migration, misplacement or breakage (5.1%), as well as death (0.4%). Side effects include, among others, memory impairment (1.1–20%), effects on cognition and psyche (10.8–33%), and depression (1.5–25%) [135]. This is especially relevant to consider, as the field of DBS has been expanding and is now used for a wide variety of disorders among others, depression, obsessive-compulsive disorder, and temporal lobe epilepsy [136–138]. It is of utmost importance to establish proper inclusion criteria to ensure that the principles of medical ethics are met [139]. These inclusion criteria are likely to be different than former protocols due to the fact that the surgery provides the most benefit early in the onset of AD, which stands in contrast with other application for DBS. Moreover, another ethical issue might be the informed consent, which can be difficult for patients suffering with cognitive impairments. Economic challenges regarding DBS must be considered as well. According to a study by Mirsaeedi-Farahani and colleagues, DBS for AD requires a success rate of 3% to produce a clinical outcome considered superior to standard treatment, for mild AD. To be considered cost-efficient a success rate of over 80% must be achieved [140].
The research in the field is growing, but key factors remain to be determined; some can be investigated by animal research (i.e., stimulation cycle, network effect), and others by clinical investigation (i.e., best response to treatment regarding disease stage, best combination of therapeutic approach for optimal improvement of quality of life).