
Abstract
Epigenetics is the study of changes in gene expression which may be triggered by both genetic and environmental factors, and independent from changes to the underlying DNA sequence—a change in phenotype without a change in genotype—which in turn affects how cells read genes. Epigenetic changes represent a regular and natural occurrence but can be influenced also by factors such as age, environment, and disease state. Epigenetic modifications can manifest themselves not only as the manner in which cells terminally differentiate, but can have also deleterious effects, resulting in diseases such as cancer. At least three systems including DNA methylation, histone modification, and non-coding RNA (ncRNA)-associated gene silencing are thought to initiate and sustain epigenetic change. For example, in Alzheimer’s disease (AD), both genetic and non-genetic factors contribute to disease etiopathology. While over 250 gene mutations have been related to familial AD, less than 5% of AD cases are explained by known disease genes. More than likely, non-genetic factors, probably triggered by environmental factors, are causative factors of late-onset AD. AD is associated with dysregulation of DNA methylation, histone modifications, and ncRNAs. Among the classes of ncRNA, microRNAs (miRNAs) have a well-established regulatory relevance. MicroRNAs are highly expressed in CNS neurons, where they play a major role in neuron differentiation, synaptogenesis, and plasticity. MicroRNAs impact higher cognitive functions, as their functional impairment is involved in the etiology of neurological diseases, including AD. Alterations in the miRNA network contribute to AD disease processes, e.g., in the regulation of amyloid peptides, tau, lipid metabolism, and neuroinflammation. MicroRNAs, both as biomarkers for AD and therapeutic targets, are in the early stages of exploration. In addition, emerging data suggest that altered transcription of long ncRNAs, endogenous, ncRNAs longer than 200 nucleotides, may be involved in an elevated risk for AD.
Keywords
INTRODUCTION
What drives the development of an organism from embryo to adult? Although at one time debated between genetic determinism and a primary influence by interactions with the surrounding environment [1], the concept of nature and nurture as being mutually exclusive [2] has evolved into a more complex and less dichotomous vision, with most biologists believing that interactions between genes and the environment steer an organism’s development. However, we have only recently begun to identify the mechanisms behind these interactions which form the basis of epigenetics [3].
Epigenetics involves changes in gene function triggered by both genetic and environmental factors, not caused by mutations in DNA sequence [4]. Such changes may relate to chromosomal ones that affect gene activity and expression, as well as heritable phenotypic changes that do not derive from genome modification. These effects on cellular and physiological phenotypic traits could be driven by external or environmental factors or be part of a normal developmental program.
Numerous central nervous system (CNS) physiological functions (neural stem cell fate determination, neural plasticity, and learning and memory, see [5]) have significant epigenetic components. This is the case also for neurodegenerative diseases. For example, in Alzheimer’s disease (AD), both genetic and non-genetic factors contribute to disease etiopathology. While over 250 gene mutations have been related to familial AD, especially those for amyloid-β protein precursor (AβPP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2), even though less than 5% of all AD cases are explained by known disease genes [6]. It is thus likely that non-genetic factors, more than likely triggered by environmental cues, are causative factors of late-onset AD. Many CNS pathologies, including AD are associated with dysregulation of DNA methylation, histone modifications, and non-coding RNAs (ncRNAs) [7]. These changes may have a significant role in AD and other neurodegenerative disorders through a variety ofpathways.
DNA METHYLATION
Methylation of DNA cytosine residues at the 5° position (5mC) is the principal and most widely characterized epigenetic modification [8]. DNA methylation is the product of a family of DNA methyltransferases (DNMTs) that add methyl groups to gene promoters to repress gene expression and block the binding of transcriptional enzymes [9]. DNMTs, in particular the subtype m5C methyltransferases (C5 Mtase), methylate the C5 carbon of cytosine in DNA to yield C5-methylcytosine [10] (see Fig. 1). C5 Mtase is particularly efficient in adding methyl groups to CpG sites, or cytosine residues in DNA directly followed by a guanidine residue on the 3′ prime side of the same DNA strand. Regions with high numbers of CpG sites, spanning at least 200 bp with higher than 60% observed-to-expected CpG ratio, are designated CpG islands and mainly occur in the gene promoter upstream of the transcription start site. Methylation of CpG islands in a gene promoter stably inhibits transcription of that gene [11].
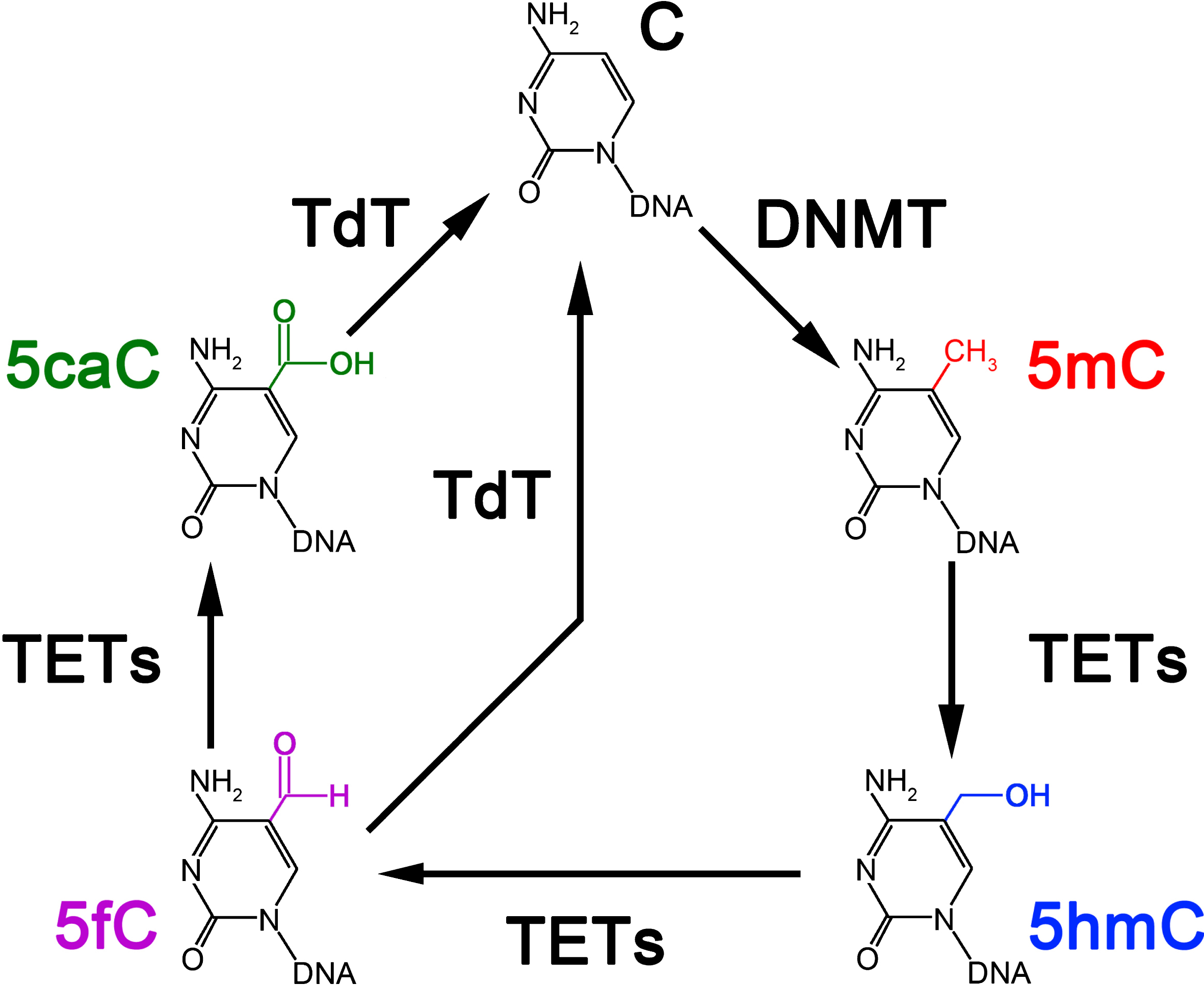
Epigenetic DNA modification. DNA methyltransferases (DNMT) add and maintain methyl groups on cytosine (C) to form 5-methylcytosine (5mC). Ten-eleven translocation (TET) family of DNA hydroxylases are responsible for executing active DNA demethylation. This process occurs through 5-hydroxymethylcytosine (5hmC) conversion to f5-formylcytosine (5fC) and 5-carboxycytosine (5caC). Both 5fC and 5caC can be converted to unmodified C by terminal deoxynucleotidyl transferase (TdT) by base excision repair.
Ten-eleven translocation methylcytosine dioxygenase 1 (Tet1) initiates DNA demethylation by converting 5-mC to 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine, and 5-carboxylcytosine at CpG-rich regions of genes [12]. Tet1 expression is high during early development and localizes to the nuclei of CNS neurons in adulthood. Studies with Tet1-knockout mice indicate its involvement in hippocampal neuronal functions, such as adult neurogenesis and memory formation and extinction [13–15]. While richly expressed in cortical and hippocampal neuron nuclei, there appears to be comparatively less Tet1 expression in glia and oligodendrocytes [13, 15]. Some authors have speculated Tet1 as an essential element for adult hippocampal neural progenitor cell proliferation, with this key regulator also influencing a number of neuronal activity-related genes [13–15].
Mammalian cells, when young, display DNA hypermethylation throughout their genome. CpG islands within the promoters of expressed genes, however, are an exception. DNA repeats extensively methylated include long interspersed nuclear elements, short Interspersed Nuclear Elements, and long terminal repeat transposable elements. In this way they remain in a constitutive heterochromatin state. Aging is accompanied by a generalized genome DNA hypomethylation, which leads to activation of normally silenced DNA sequences. It bears mentioning that DNA methylation levels of some CpGs are age-related allowing the estimation of DNA methylation age based on the cumulative assessment of CpGs [16]. DNA methylation may also increase in a non-stochastic manner over the CpG islands of certain genes, correlating with their heterochromatinization and silencing. In spite of conflicting results [17], the majority of global methylation studies employing antibody-specific immunohistochemistry on postmortem AD brain tissue show a reduction in DNA methylation in temporal neocortex [18], entorhinal cortex [19], and hippocampus [20]. A reduction in global DNA methylation could have a role in activation of microglia and astrocytes and output of pro-inflammatory cytokines during aging [21], with the potential for creating a vicious feedback cycle.
Epigenetic processes play a role in deregulation of gene expression that occurs in AD. Initially focusing on AβPP gene methylation, findings from these studies proved relatively conflicting and inconsistent, possibly due to different techniques, limited sample numbers, and the use of heterogeneous tissue. Other AβPP-related genes (apolipoprotein E, nicastrin and beta-site AβPP Cleaving Enzyme 1) were found to be unchanged in AD brain samples. However, Di Jager et al. [22] functionally validated CpG associations significantly associated with the burden of AD pathology and identified the nearby genes whose RNA expression was altered in AD: ANK1, CDH23, DIP2A, RHBDF2, RPL13, SERPINF1, and SERPINF2. These authors suggest that such DNA methylation changes may have a role in the onset of AD, given that they were seen in pre-symptomatic subjects and that six of the validated genes connect to a known AD susceptibility gene network.
Alterations in DNA methylation are found in a number of genes not directly related to amyloid and tau. The methylation process occurs also in normal aging, with hyper- or hypo-methylation appearing to be faster in AD. Because methylation takes place mainly in CpG islands, and about 70% of promoters in man contain at least one CpG island [23]; the presence of multiple methylated CpG sites in the promoters of these islands causes stable silencing of genes [24, 25]. On the other hand, a decrease in methylation at CpG sites leads to gene activation [26] (Table 1).
A selection of genes in AD patients subject to methylation (inhibiting) and demethylation (activating) processes
5hmC, 5-hydroxymethylcytosine. The first oxidative product in the demethylation of 5-methylcytosine (see Fig. 1).
enlargethispage 1pt
HISTONE MODIFICATIONS
Histones are nuclear proteins enwrapped by DNA to form a nucleosome. The five major families of histones are: H1/H5, H2A, H2B, H3, and H4. Histones H2A, H2B, H3, and H4 are considered core histones, withH1/H5 as linker histones. Histone protein tails have residues capable of being acetylated, methylated, phosphorylated, ubiquitylated, and sumoylated. Post-translational histone modifications, such as methylation and acetylation of lysine residues on histone tails affect gene expression, mainly by altering chromatin structure: euchromatin, a lightly packed form of chromatin enriched in genes allows gene transcription, whereas heterochromatin, a tightly packed form of chromatin, blocks it. Furthermore, there are enzymes that add modifications, such as histone methyltransferases, and others that remove these modifications, including histone demethylases. A complex interplay between activating and repressing histone modifications is a major regulatory pathway for gene expression. It should be noted that lysine methyltransferases have a broad spectrum of non-histone protein substrates [27].
Methylation
Methylation of histone tails results in either increased or reduced gene expression, depending on the amino acid methylated. For instance, H3K4 tri-methylation [28] is associated with increased transcription, whereas H3K9me3 results in transcription repression [29].
Acetylation
Unlike methylation of histone tails that either induces or represses gene transcription, acetylation of histone tails is solely associated with active gene expression. Histone acetylation is currently the most researched epigenetic mechanism in CNS dysfunction and neurodegenerative diseases as it relates to astrocyte function. Conditioned medium from lipopolysaccharide (LPS)-stimulated microglia decreased total H3 and H4 acetylation in cultured astrocytes and increased astrocyte cell death from H2O2 treatment, while decreasing expression of the anti-oxidant factor nuclear factor erythroid 2–related factor 2 [30]. Valproic acid, a histone deacetylase (HDAC) inhibitor, reportedly attenuates the detrimental effects of conditioned medium from LPS-treated microglia [31].
The ATP-binding cassette sub-family A member 7 (ABCA7) gene
ABCA7 codes for a member of the superfamily of ATP-binding cassette (ABC) transporters, whose function being to carry a variety of molecules across extra- and intra-cellular membranes [151]. ABC genes comprise seven subfamilies: ABC1, MDR/TAP, CFTR/MRP, ALD (adrenoleukodystrophy), OABP, GCN20, and White. ABCA7 belongs to the ABC1 subfamily, whose members are the only major ABC subfamily exclusive to multicellular eukaryotes. ABCA7 is found mainly in myelo-lymphatic tissues, with greatest expression in peripheral leukocytes, spleen, thymus, and bone marrow. This transporter’s function remains unknown, although its expression pattern points to a role in lipid homeostasis in immune system cells. Two transcript variants result from alternative splicing ABCA7, both predisposing to AD [152]. Indeed, the Icelandic database of Decode Genetics points to a two-fold greater risk of developing AD when ABCA7 inactive variants of are present.
Deacetylation
Histone tail deacetylation antagonizes gene expression at loci throughout the genome [32]. Good evidence points to histone acetyltransferase and HDACs involvement in various cognitive processes, proposing that dynamic regulation of histone acetylation status is associated with synaptic plasticity and memory [33, 34]. Among histone-modifying enzymes, HDAC2 is a critical negative regulator of structural and functional plasticity in the mammalian nervous system [35]. HDAC2 localizes to the promoters of numerous synaptic-plasticity-associated genes, where it deacetylates histone substrates [36]. Loss of HDAC2 or treatment with HDAC inhibitors promotes synaptic gene expression, long-term synaptic plasticity, memory processes, and decreases disease progression both in AD subjects and in a transgenic mouse AD model [37], while HDAC2 over-expression has the opposite effects [36–39].
Modified HDAC function can contribute to numerous pathological states, including cancer, cardiovascular disease, and neurological diseases [40]. HDAC2 levels are elevated in AD brain and in mouse AD models [41]. HDAC2 upregulation in the latter results from both transcriptional and post-transcriptional mechanisms and contributes to cognitive impairment; HDAC inhibitors and HDAC2 knockdown result in striking recovery of impaired cognitive functions [36]. As such, HDAC2 is a promising target for treating the cognitive symptoms of AD and other neurological disorders.
Sp3 is a gene that encodes transcription factors which bind to consensus GC- and GT-box regulatory elements in target genes. Functional screening of potential co-regulators revealed that knockdown of Sp3 was similar to HDAC2 knockdown in terms of facilitating synaptic transmission. Consistent with a role in recruitment of HDAC2 to target genes, knockdown of Sp3 reduced HDAC2 occupancy and increased histone acetylation of synaptic gene promoters and facilitated synaptic gene expression. As with HDAC2, Sp3 expression was elevated in brain of a mouse AD model and in AD patients. Expression of an HDAC2 fragment containing the Sp3-binding domain counteracted the synaptic plasticity and memory defects found in a mouse AD model. Together, these findings indicate that HDAC2 and Sp3 cooperate to regulate neuronal plasticity genes and provide proof-of-principle that disruption of the HDAC2-Sp3 interaction is an effective strategy to disrupt the synaptic plasticity-suppressingfunctions of this complex [42].
Phosphorylation, ubiquitylation, and SUMOylation
Histone phosphorylation is usually associated with DNA damage, with increased phosphorylation of histone H2AX around the sites of DNA damage in hippocampal and cortical astrocytes in AD patients [43]. Increased phosphorylation of H3 histone is found in frontal cortex of AD and bipolar disorder patient brains [44]. Furthermore, Ogawa et al. [45] described, in AD hippocampal neurons, co-localization between tangles and hyperphosphorylation of H3 Ser10. Unlike the case for actively dividing cells, phosphorylated histone H3 was restricted to the neuronal cell cytoplasm (and not nucleus) despite activation of the mitotic machinery. Therefore, the aberrant cytoplasmic localization of phosphorylated histone H3 may be a sign of mitotic disbalance that leads to neuronal cell dysfunction and neurodegeneration in AD. Histone phosphorylation appears to be part of a complex interplay between other epigenetic markers, such as histone acetylation and methylation, and DNA methylation. Indeed, histone phosphorylation has been demonstrated to increase pro-inflammatory gene activation [46, 47].
Histone ubiquitylation is another consequence of DNA damage. H2A ubiquitylation usually leads to gene silencing, whereas H2B ubiquitylation has been linked to gene activation. Histone ubiquitylation is important for regulating stem cell maintenance and differentiation, including neural stem cells [48]. Little is known, however, role for histone ubiquitylation in neurodegenerative diseases.
Small Ubiquitin-like Modifier (SUMO) proteins are a family of small proteins capable of modifying the function of other proteins through covalent interactions. SUMOylation is a post-translational modification that participates in nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, stress response, and cell cycle progression [49]. SUMOylation involves an enzymatic cascade analogous to that in ubiquitination. Unlike the case for ubiquitin, SUMOylation does not tag proteins for degradation. Rather, it can repress gene transcription through the opposing actions of histone acetylation and ubiquitylation, and reduction of histone SUMOylation leads to higher levels of acetylation [50]. Although post-translational protein SUMOylation has been suggested to contribute to pathogenesis of several neurodegenerative diseases, such as AD, data are limited in these cases. A major finding concerning SUMOylation in neurodegeneration was reported by Tao et al. [51], who demonstrated that SUMOylation of HDAC1 was an endogenous protective mechanism against amyloid-β (Aβ) toxicity in a mouse AD model. These authors also reported that acute Aβ treatment in rats induced expression of a protein inhibitor of signal transducer and activator of transcription 1 (PIAS1), which is a SUMO E3 ligase [51].
A number of proteins involved in AD pathology (AβPP, tau, β-site amyloid precursor protein cleaving enzyme 1 (BACE1), glycogen synthase kinase-3β, and c-Jun N-terminal kinase) are SUMO targets [52]. Furthermore, AD patients have altered levels of SUMOylation and SUMO-related protein expression. For example, SUMO3 labeling was increased in postmortem AD hippocampus [53]. In confirmation of these results, genomic analysis of Korean patients affected by late-onset AD showed mutations of the Ubc9 gene (UBE2I), suggesting a link with increased risk of developing AD for this population [54].
SUMO ligases, including SUMO-1 and PIAS1, are expressed in rat CNS, and decrease with age or exposure to LPS [55]. These authors also reported that SUMOylation of the transcription factor CCAAT/enhancer-binding protein regulates expression of the gene for inducible nitric oxide synthase; decreased SUMOylation led to increased inducible nitric oxide synthase expression in cultured astrocytes. Hoppe et al. [56] found that Aβ1 - 42-treated mouse astrocytes contained reduced levels of SUMO-1-conjugated proteins, with over-expression of constitutively active SUMO-1 halting some of the morphological effects of Aβ1 - 42. Similarly, SUMOylation of liver X receptors in cultured astrocytes, by the SUMO ligases PIAS1 and HDAC4, reduced interferon gamma induced activation of signal transducer and activator of transcription 1 and subsequent inflammatory responses [57]. These results indicate that SUMOylation is involved in gene expression and that SUMOylation can reduce inflammation, which points to a potential role for SUMOylation in neurodegeneration [58, 59].
NON-CODING RNAs
Non-coding DNA sequences are DNA components that do not encode protein sequences. Some non-coding DNA is transcribed into functional ncRNA species (transfer RNA, ribosomal RNA, and regulatory RNAs). Non-coding DNA also regulates the transcription and translation of protein-coding sequences, scaffold attachment regions, centromeres, and telomeres. The advent of RNA sequencing has led to the discovery of new classes of RNA [60]. While known ncRNAs number in the thousands, their roles in the regulation of gene expression and genomic organization remain in their infancy. Among the classes of ncRNA, microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have received the most attention. The former have with a well-established regulatory role [61, 62], while the latter appear to be involved in the development of a number of human diseases [63–65]. The present discussion will be restricted to the role of miRNAs and lncRNAs in AD.
miRNA
MicroRNAs are small (20–24 nucleotide) ncRNAs that, although coding for proteins, are able to alter gene expression [66]. MicroRNAs prevent translation by binding to the 3′ untranslated end of mRNA, resulting in mRNA degradation [67]. MicroRNAs thus represent another form of epigenetic regulation by altering translation. MicroRNAs, which are highly expressed in CNS neurons, play a major role in neuron differentiation, synaptogenesis, and plasticity. MicroRNAs profoundly impact higher cognitive functions, as evidenced by their functional impairment being involved in the etiology of neurological diseases, including AD [68]. A growing body of evidence points to alterations in the miRNA network as active contributors to AD disease processes [69; for a recent review see 70].
miRNA Regulation of Aβ
AD pathology is associated with a number of altered features of AβPP: increased expression, polymorphisms in its promoter, abnormal processing, and altered Aβ clearance [71]. MicroRNAs regulate expression of AβPP and other enzymes involved in Aβ processing, in particular BACE1.
AβPP: miR-20a, miR-19, and miR-106a/b (all miR-20a family members) [72] and miR-101, among others, directly regulate APP mRNA in cultured human cells [73]. miRNAs are also involved in regulating alternative splicing of AβPP. In AD patients, AβPP in neurons undergoes alternative splicing at exons 7, 8, and 15, with increased expression levels in exons 7 and/or 8 isoforms of AβPP [74]. In addition, abnormal splicing of AβPP in neurons was associated with heightened Aβ production [75]. On the other hand, miR-124 was downregulated in AD brains [76].
BACE1: Increased BACE1 expression and activity have been reported in sporadic AD brain [77]. The miR-29 family (homo sapiens (hsa)-miR-29a, hsa-miR-29b, and hsa-miR-29c) has received much attention in the context of BACE1, with several reports linking this family to BACE1 regulation in vivo and in cultured cells. MicroR-29 family members show marked reductions in AD brain, and specifically so in relation to AD dementia. These reductions are associated with exceptionally high BACE1 protein, and loss of miR-29a/b-1 cluster suppressing activity in cultured human cells results in increased Aβ production [78]. miR-339-5p, miR-195 [79], and miR-107 [80], among other miRNAs, are deregulated in AD brain and can directly target BACE1 in vitro.
miRNA regulation of tau
The microtubule-associated protein tau stabilizes microtubule structure and facilitates axonal transport. In AD, tau undergoes translocation to the somato-dendritic compartment, where it undergoes hyperphosphorylation and misfolding to generate neurotoxic intracellular aggregates (neurofibrillary tangles) [81]. Tau pathology may be a consequence of an alteration in one or more processes regulating its metabolism, e.g., expression, localization, transcriptional/posttranslational modification, and clearance. Hyperphosphorylation may follow from upregulation and/or abnormal expression of tau kinases, downregulation of phosphatases, mutation, and covalent modification of tau [82, 83]. The following two sections discuss miRNA involvement in regulating tau metabolism in pathological conditions.
Tau mRNA expression and metabolism: The miR-132/miR-212 cluster has been tied to the regulation of tau expression. Decreased levels of miR-132 levels are reported in advanced stage AD brain [57, 85]. Other miRNAs involved in tau expression and metabolism include: miR-219, that represses tau synthesis post-transcriptionally and is downregulated in autopsy brain tissue taken at autopsy from AD patients and subjects with severe primary age-related tauopathy [86]; miR-128a, that modulates BAG2 expression, a co-chaperone suggested to be involved in tau aggregation and degradation [87].
Tau phosphorylation: Neurofibrillary tangles in AD brain are composed mainly of hyperphosphorylated microtubule-associated proteins, whose abnormal phosphorylation may represent a key process leading to their aggregation [88]. The state of tau phosphorylation represents a fine balance between kinases and phosphatases, processes that may be regulated by miRNAs. Interestingly, hyperphosphorylation of tau at pathological sites was accompanied by increased phosphorylation of mitogen-activated protein kinase 3/extracellular signal-regulated kinase 1. In mouse neurons: extracellular signal-regulated 1 is downregulated by members of the miR-15 family (including miR-15a), while this miRNA is reduced in AD brain [89]. Glycogen synthase kinase-3β, also known as tau protein kinase, occupies a key position in Aβ production and neurofibrillary tangle formation and is regulated, at least in smooth muscle cells, by miR-26a [90, 91]. Together with miR-26b, miR-26a appears critical for brain-derived neurotrophic factor expression [92]. Postmortem AD brain reportedly contains markedly raised miR-26b levels in the temporal gyrus of [93]. miR-922 is another miRNA that increases tau phosphorylation. In human cell lines, Cogswell et al. [94] showed that miR-922 heightening tau phosphorylation by downregulating UCHL1, a deubiquitinating enzyme that is decreased in AD brain. Furthermore, UCHL1 levels were inversely proportional to tangle number [95]. Intriguingly, converging evidence suggests that psychotic AD (AD+P) [96] is associated with an acceleration of frontal degeneration, with tau pathology playing a primary role [97]. Within this context, a new study by Kwon et al. [98] shows that Tet1 over-expression leads to anxiety-like behavior and enhanced fear memories via the activation of calcium-dependent cascade through Egr1 expression in mice.
miRNA regulation of lipid metabolism
The apolipoprotein E gene ɛ4 allele is probably the strongest genetic risk factor known to-date for AD [99]. It is associated with modification in the metabolism of a number of lipids linked to AD [100]. Cholesterol metabolism is likely to be the main player, with dysregulation of genes involved in its biosynthesis/efflux being associated with AD evolution. miR-33, by regulating lipid metabolism via inhibition of ABCA1, can influence AD. ABCA1 plays a direct role in AD by decreasing levels of Aβ [102]. miR-33 directly regulates ABCA1 in human neuronal cell lines and in mouse primary neurons and astroglia [103]. This has practical implications for AD: Kim et al. [104] demonstrated that miR-33, by downregulating ABCA1, affects Aβ levels. However, deregulation of miR-33 in AD brain remains to be proved. ABCA1 and cholesterol metabolism are regulated also by miR-758. It has been proposed that this miRNA is implicated in the regulation of AD development [105]. A number of other miRNAs (miR-137, miR-181c, miR-9, and miR29a/b) have been linked to impaired lipid metabolism in AD. For a recent review see Goedeke and Fernández-Hernando [99].
Biomarker assay in blood, plasma, and CSF (analyzed by ROC)
CSF, cerebrospinal fluid; MCI, mild cognitive impairment; MMSE, Mini-Mental State Exam; qPCR, quantitative Real Time polymerase chain reaction; ROC, receiver operating characteristic.
miRNA regulation of neuroinflammation
In AD, as with other neurodegenerative diseases, binding of misfolded/aggregated proteins to glial cell receptors triggers innate immune responses that contribute to disease progression. Genes that regulate glia clearance of misfolded proteins and provoke inflammatory reactions appear to increase one’s risk for AD [106]. Nuclear factor-kappaB (NF-κB), an immune and stress-induced transcription factor, regulates transcription of several miRNAs linked to neuroinflammation. Under physiological conditions, NF-κB activity induced by inflammation is blocked by acetylcholine [107]. At the same time, production of acetylcholine is markedly compromised in AD [108]. Consequently, miR-34a, a miRNA controlled by NF-κB, is upregulated in hippocampal CA1 region of AD brain [109]. These same authors showed that miR-34a regulates, in mouse microglia, triggering receptor expressed on myeloid cells-2, a critical factor for Aβ1 - 42 clearance that is downregulated in AD brain CA1 region [110, 111]. This link is strongly supported by the observation that rare heterozygous variants of triggering receptor expressed on myeloid cells-2 are associated with a significant increase in AD risk [112], miR-146a [113], miR-155 [114], and miR-181 family [115], also involved with the innate immune system, are known to be relevant to AD.
Long non-coding RNAs (lnc-RNAs)
LncRNAs are endogenous, non-coding RNAs longer than 200 nucleotides [116]. Transcriptome assembly after next-generation RNA sequencing has revealed more than 50,000 lcnRNAs in various human tissues. LncRNAs are transcribed from introns, exons of protein-coding genes, and from intergenic regions [117, 118]. LncRNAs are transcribed as precursor transcripts, after which they are subject to splicing, maturation, exportation, and decay in analogy to mRNAs but, and unlike the latter, lncRNA genes are poorly conserved [119]. LncRNA binding to nucleic acid or protein regulates gene expression, cell metabolism, division, differentiation, survival, senescence, neurodegeneration, and aging [120–125]. LncRNAs present different structures, including mRNA-like forms and circular RNAs [117]. LncRNAs may comprise just 100 kilobase pairs [126], and possess fewer long exons, although these are shorter than mRNAs [119, 127].
Altered lncRNA transcription may be involved in increasing the risk of AD. There is evidence for several specific lncRNAs being dysregulated in AD, including AβPP, and BACE1 [128]. Such altered expression is detectable not only in AD brain, but also in the plasma and CSF of these patients [128–131]. In particular, BACE1-AS lncRNA is an antisense RNA transcribed by RNA polymerase II from the complementary strand of BACE1 in the locus 11q 23.3 [132]; it is directly implicated in the increased abundance of Aβ1 - 42 in AD. BACE1-AS stabilizes BACE1 mRNA by forming an RNA duplex to mask the binding site [132], thereby preventing BACE1 degradation by MiR-485-5p [133]. Higher BACE1-AS levels can increase BACE1 protein and elevated levels of BACE1 inactivate the cAMP-protein kinase A-cyclic AMP response element binding protein pathway, leading to deficits in learning and memory, and to excessive production of Aβ1 - 42 and amyloid plaques [74].
A number of other lncRNAs are been identified in brain that are related to AD. 51A is an antisense RNA transcribed from intron 1 of the Sorl1 gene. Sorl1 is reduced in AD brain [134], and a loss of Sorl1 function disrupts AβPP cleavage and promotes an increase of neurotoxic Aβ peptides [135]. Increased expression of 51A lncRNA leads to a strong decrease of the Sorl1 splice variant A mRNA and, resulting in increased amounts of Aβ1 - 40 and Aβ1 - 42. BC200 is a small untranslated RNA, transcribed by RNA polymerase III in the neuronal cell body where it is transported to dendrites as ribonucleoprotein particles. BC200 has been implicated in protein synthesis. Through its binding to the poly(A)-binding protein 1, BC200 takes part in regulating transcription initiation. Higher levels of BC200 in brain are associated with the severity of AD [136, 137]. GDNFOS is a cis-antisense transcript from the glial cell line-derived neurotrophic factor (GDNF) gene containing four exons that can give rise to three spice variants (GDNFOS1, GDNFOS2, and GDNFOS3). The first two splice variants are non-coding RNAs, but GDNFOS3 possesses a potential open reading frame. While conceivable that GDNFOS3 may influence GDNF concentration in the brain, more evidence will be needed to confirm a interaction between GDNFOS and GDNF mRNA [136]. LncRNA 17A is an antisense lncRNA complementary to an intronic region of the GABBR2 gene, expressed in neuroblastoma, and leads to an increase in Aβ secretion. Other lncRNAs implicated in AD are NAT-Rad18, Uchl1 RNA (an antisense lncRNA complementary to mouse ubiquitin carboxy-terminal hydrolase L1) [138], CDKN2B-AS (ANRIL, antisense non-coding RNA in the INK1 locus) [139], HARIF and HARIR located in the highly accelerated region 1 (HAR1) [140, 141], Sox2ot (a RNA that overlaps SOX2 gene), and Snhg3 (a lncRNA associated to ribosomal processing) [142]. In addition, reassessment of microarray data using a novel algorithm revealed about 100 brain-specific intergenic lncRNAs that showed changes greater than two-fold in AD [131, 143].
CONCLUSIONS
A rapidly growing body of data associates epigenetic alterations to the development of neuropathological and neurodevelopmental disorders [144] Conceivably, disruption of gene expression due to epigenetic changes throughout the human lifespan could, over the long term, act as a ‘seed’ for development of disorders [145]. Collectively, the studies described here point to a role for epigenetic mechanisms in the complex etiology of AD. As such, understanding epigenetic dysregulation in AD could contribute to our view of the origin and progression of AD and, possibly, the development of efficacious therapeutics. However, one caveat with epigenetic studies is the issue of causality. In other words, alterations in DNA modifications could be either causal to the disease process or could themselves arise as a consequence of disease-associated pathological changes. One way to overcome such doubts would be to determine if a given epigenetic variation was present before the appearance of disease symptoms, preferably via longitudinal studies following the same cohort of individuals over a number of years (an approach that, in itself, is not only very costly but also requires a protracted time frame).
Could there be a disconnect between epigenetic variation which arises before disease onset and actual disease causation? For instance, while aging is accompanied by a generalized genome DNA hypomethylation, not all old people with DNA hypomethylation develop AD. Clearly, the question of cause and effect of aging and pathology awaits a conclusive answer. Given the potential reversibility of epigenetic changes, identifying disease-causative mechanisms may one day provide viable therapeutic targets.
Given the failure of AD clinical trials to date, focus is now shifting to diagnose AD at as early a stage as possible, even before onset of cognitive decline. Despite the inherent difficulties, timely disease detection offers a multitude of benefits, not the least of which are opportunities for early intervention, better management of symptoms, and cost savings [146]. miRNAs have emerged as potential candidates for reliable biomarkers of early-stage AD, being present in biofluids and displaying high stability in terms of storage/handling [147] Table 2 summarizes such studies in which receiver operating characteristic (ROC) curve analysis was applied to optimize biomarker validity. Recent attempts at using miRNAs in serum, plasma, and cerebrospinal fluid (CSF) as AD biomarkers have identified a number of differentially expressed miRNAs [cf. 148], although lack of consistency between studies was evident. miR-181c emerged as one of only a few miRNAs identified in multiple studies, being downregulated in AD CSF and serum [80], along with miR-146a (involved in innate immune response) upregulated in AD brain and CSF [149]. Moreover, ncRNAs, miRNAs—and especially lncRNAs—as therapeutic targets are only beginning to be considered. Even so, these transcripts represent potential targets for two reasons: 1) lncRNA expression seems to be rather cell- and tissue-specific [150]; 2) the sequence-specific function of lncRNA can be advantageous in designing specific therapies. As a closing, albeit speculative thought, is the possibility that epigenetic mutations exist that function to protect or promote recovery from the neuropathological sequelaeof AD.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0259r2).