
Abstract
Trans-active response DNA-binding protein of 43 kDa (TDP-43) is a highly conserved and ubiquitously expressed nuclear protein. As a member of heterogeneous ribonucleoproteins, TDP-43 plays pivotal roles in mRNA processing. We recently found that TDP-43 promoted tau mRNA instability via acting on the 3’-untranslated region of its mRNA and enhanced tau exon 10 inclusion. TDP-43 is a phospho-protein. The function and the pathological aggregation of TDP-43 are regulated by the phosphorylation. In the present study, we determined phosphorylation of TDP-43 by cyclic AMP-dependent protein kinase (PKA). We found that TDP-43 was co-immunoprecipitated by and co-localized with PKA in the nucleus. PKA phosphorylated TDP-43 at Ser379, Ser403/404, and Ser409/410 in vitro and in cultured cells. Phosphorylation of TDP-43 at these sites enhanced mutually their phosphorylation by PKA in vitro and in cultured cells. Overexpression of PKA suppressed TDP-43’s activity in promoting tau mRNA instability and tau exon 10 inclusion. These findings shed light on the role of PKA in phosphorylation and function of TDP-43. Downregulation of PKA signaling in AD brain may attenuate the impact of TDP-43 pathology in tau pathogenesis.
INTRODUCTION
Trans-active response DNA-binding protein of 43 kDa (TDP-43) is a highly conserved and ubiquitously expressed nuclear protein involved in the regulation of gene transcription and RNA metabolism, including mRNA stability, alternative splicing, and transport to cytoplasm [1–3]. TDP-43 is deposited as hyperphosphorylated cytoplasmic inclusions in the brains of patients with amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U), now referred to as TDP-43 proteinopathies [4]. In addition, TDP-43 pathology is concomitant with Alzheimer’s disease (AD) and other tauopathies like cortical basal degeneration [5–7].
Tau is a major neuronal microtubule associated protein which is hyperphosphorylated and aggregated into neurofibrillary tangles in the brain of AD and related tauopathies [8–10]. Six tau isoforms are expressed from a single gene by alternative splicing of exons 2, 3, and 10 of its pre-mRNA in adult human brain [11]. Tau exon 10 encodes the second microtubule binding repeat [12], and its alternative splicing generates tau isoforms with three or four microtubule-binding repeats (3R-tau or 4R-tau). 3R-tau and 4R-tau level is equal in normal adult human brain [12]. Alteration in 3R-tau/4R-tau ratio has been seen in many neurodegenerative disorders, including frontotemporal dementia, Pick’s disease, corticobasal degeneration, or progressive nuclear palsy [13–17]. We recently found that TDP-43 promotes tau mRNA instability and enhances tau exon 10 inclusion, leading to suppression of tau expression and increase the ratio of 4R-tau/3R-tau [18, 19]. Cytoplasmic aggregation of TDP-43 may lead to its depletion from the nucleus, resulting in increase of 3R-tau and total tau expression.
Cyclic AMP (cAMP)-dependent Ser/Thr protein kinase (PKA) is a key kinase that phosphorylates many proteins involved in the etiology of AD as well as other tauopathies [20]. PKA is an inactive tetrameric holoenzyme consisting of two catalytic subunits (PKAc) and two regulatory subunits in the absence of cAMP. There are three isoforms, α, β, and γ, of PKA catalytic subunit, α and β are expressed ubiquitously [21]. Stimulation by cAMP dissociates the holoenzyme and causes a translocation of a fraction of PKAc into the nucleus to phosphorylate nuclear proteins. Several studies reported that PKA phosphorylates splicing factors and modulates the pre-mRNA splicing, including tau pre-mRNA [22–26]. However, whether PKA phosphorylates TDP-43 and affects its function are unknown. In the present study, we found that PKA phosphorylated TDP-43 and modulated its function in the regulation of tau mRNA stability and tau exon 10 splicing.
MATREIALS AND METHODS
Plasmids, antibodies, and other reagents
pCI/PKAcα, pCI/PKAcβ tagged with hemagglutinin (HA) and Flag, pCI/TDP-43 tagged with HA, mutations of TDP-43 at Ser409/410 to Ala (TDP-43S409/410A) or to Asp (TDP-43S409/410D), pEGFP/tau 3’-untranslated region (3’-UTR) were constructed as described previously [10, 27]. pcDNA/TDP-43·myc·his was generated by polymerase chain reaction (PCR) amplification from pCI/TDP-43 and confirmed by DNA sequence analysis. Mutations of TDP-43 at Ser379 or Ser403/404 to Ala (TDP-43S379A or TDP-43S403/404A) or to Asp (TDP-43S379D or TDP-43S403/404D) were achieved by using the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). pCI/SI9-LI10 containing a tau mini-gene, SI9-LI10, comprising tau exons 9, 10, 11, and part of intron 9 and the full-length of intron 10 was a gift from Dr. Jianhua Zhou of the University of Massachusetts Medical School [28]. Monoclonal anti-HA and polyclonal anti-HA were bought from Sigma (Sigma, St Louis, MO, USA). Polyclonal anti-TDP-43 was from Cell signaling technology (Danvers, MA, USA). Polyclonal anti-PKAcα and anti-PKAcβ, siRNAs of PKAcα and PKAcβ were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal anti-pS379-TDP-43, anti-pS403/404-TDP-43 and anti-pS409/410-TDP-43 were made by Abmart company (Shanghai, China). Peroxidase-conjugated anti-mouse and anti-rabbit IgG were from Jackson Immuno Research Laboratories (West Grove, PA, USA). Alexa 488-conjugated goat anti-rabbit IgG, Alexa 555-conjugated goat anti-mouse IgG and TO-PRO-3 iodide (642/661) were from Invitrogen (Invitrogen, Carlsbad, CA, USA). [γ-32P]-ATP was from MP Biomedicals (Irvine, CA).
Cell culture and transfection
Human embryonic kidney cell line (HEK-293FT and HEK-293T) and human cervix epithelia cell line (HeLa) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA), mouse neuroblastoma N2a cells were maintained in DMEM/F12 supplemented with 10% FBS at 37°C (5% CO2). All transfections were performed with FuGENE HD (Roche Diagnotics, Indianapolis, IN, USA) or Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Immunofluorescence
HeLa cells were plated in 24-well plate onto glass coverslips 1 day before transfection at around 30% confluence. pcDNA/TDP-43·myc·his was transfected with pCI/PKAcα or pCI/PKAcβ for 48 h, then the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min at room temperature. After washing with PBS, the cells were blocked with 10% goat serum in 0.2% Triton X-100 in PBS for 2 h at 37°C, and then incubated with monoclonal anti-myc (1:200) and polyclonal anti-HA (1:500) overnight at 4°C. The cells were washed and incubated for 2 h with Alexa 488-conjugated goat anti-rabbit IgG and Alexa 555-conjugated goat anti-mouse IgG (1:500) plus TO-PRO-3 iodide (5μg/ml) at room temperature. After washing with PBS, the cells were mounted with anti-fade mounting media (Roche, Diagnostics, USA), and observed with a Leica TCS-SP2 laser scanning confocal microscope.
Immunoprecipitation and co-immunoprecipitation
For immunoprecipitation (IP), TDP-43, TDP-43S379A, TDP-43S403/404A, TDP-43S409/410A, or PKAcα, PKAcβ was overexpressed separately in HEK-293FT cells. The cells were washed with PBS and lysed in buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.2% sodium deoxycholate, 0.1% NP-40, 0.1% Triton X-100, 1 mM Na3VO4, 50 mM NaF, 2 mM EDTA, 1 mM AEBSF, and 10μg/ml each of aprotinin, leupeptin and pepstatin) on ice for 20 min, then centrifuged at 15,000× g, 4°C for 5 min. The supernatant was incubated with monoclonal anti-HA pre-coupled protein G beads overnight at 4°C.
For co-immunoprecipitation (co-IP), N2a cells were washed twice with PBS, and lysed by sonication in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM Na3VO4, 50 mM NaF, 2 mM EDTA, 1 mM AEBSF, and 10μg/ml each of aprotinin, leupeptin, and pepstatin), then centrifuged at 15,000×g, 4°C for 5 min. The extract was incubated with protein G beads pre-coupled with anti-PKAcα or anti-PKAcβ overnight at 4°C.
The beads were washed with lysis buffer twice and with Tris-buffered saline (TBS) twice, then the bound proteins were eluted by boiling in 2×Laemmli sample buffer and subjected to western blots or used for in vitro phosphorylation assay.
In vitro phosphorylation of TDP-43 by PKA
Immunopurified TDP-43, TDP-43S379A, TDP-43S403/404A or TDP-43S409/410A was incubated with IP-PKAcα or IP-PKAcβ in a reaction buffer consisting of 50 mM HEPES, pH6.8, 10 mM MgCl2, 10 mM β-mercaptoethanol, 1 mM EGTA, and 0.2 mM [γ-32P] ATP (500 cpm/pmol). After incubation at 30°C for 1 h, the reaction was stopped by boiling with an equal volume of 2×Laemmli sample buffer. The reaction products were separated by SDS-PAGE or analyzed by western blots with indicated antibodies. Incorporation of 32P was detected by exposure of the dried gel to phosphor-image system (BAS-1500, Fuji film).
Knockdown of PKA with RNA interference
HEK-293FT cells were transfected with short interfering RNAs, siPKAcα and siPKAcβ (siPKAcα, a pool of 3 siRNAs including sc-36240A: Sense: CCAUGAAGAUCCUCGACAAtt and Antisense: UUGUCGAGGAUCUUCAUGGtt; sc-36240B: Sense: CCAUCCAGAUCUAUGAGAAtt and Antisense: UUCUCAUAGAUCUGGAUGGtt; sc-36240C: Sense: GAGUAACUUUGACGACUAtt and Antisense: UAGUCGUCAAAGUUACUCGtt; siPKAcβ, a pool of 3 siRNAs including sc-39158A: Sense: GAAGAGUCAUGUUGGUAAAtt and Antisense: UUUACCAACAUGA CUCUUCtt; sc-39158B: Sense: CUCAGAUAGUGCUAA CAUUtt and Antisense: AAUGUUAGCACUAUCUGAGtt; sc-39158C: Sense: GCAUUAGGAGUGCUAAUCUtt and Antisense: AGAUUAGCACUCCUAAUGCtt) using Lipofectamine 2000 according to the manufacturer’s instructions. The same amount of scramble siRNA was used for control. After 48 h transfection, RNA was extracted as described below.
Reverse transcription-PCR (RT-PCR) and Real-time quantitative PCR (qRT-PCR)
Total cellular RNA was extracted from cultured cells by using the RNeasy mini kit (Qiagen, GmbH, Germany) according to the manufacturer’s instruction. One microgram of total RNA was used for first-strand cDNA synthesis with oligo-(dT)18 by using the Omniscript reverse transcription kit (Qiagen). PCR was performed by using PrimeSTARTM HS DNA Polymerase (Takara Bio Inc., Otsu, Shiga, Japan) with forward primer 5’-GGTGTCCACTCCCAGTTCAA-3’ and reverse primer 5’-CCCTGGTTTAT GATGGATGTTGCCTAATGAG-3’ to measure the alternative splicing of tau exon 10. The conditions of PCR were: 98°C for 5 min, 98°C for 10 s, 68°C for 40 s for 30 cycles, and then 68°C 10 min for extension. The PCR products were resolved on 1.5% agarose gel, visualized by ethidium bromide staining and quantitated using the Molecular Imager system (Bio-Rad).
For qRT-PCR, total cellular RNA was extracted and then qPCR assay was performed in a final volume of 25μl containing 2×EvaGreen qPCR master mixture 12.5μl (Agilent Technologies, Santa Clara, CA, USA), 1μl DNA template, 0.5μl (10μM) of forward and reverse primers, and 0.375μl of dye (1:500). The qPCR was performed using these primers: GFP (forward, 5’-TGAACCGCATCGAGCTGAAGGG-3’; reverse, 5’-ACCTTGATGCCGTTCTTCTGCTTG-3’) and human GAPDH (forward, 5’-CATGAGAAGT ATGACAACAGCCT-3’; reverse, 5’-AGTCCTT CCACGATACCAAAGT-3’). Amplification was conducted in a MX3000P real-time PCR system (Stratagene), and the PCR conditions were 95°C for 10 min, 95°C for 30 s, 55°C for 1 min, 72°C for 1 min for 40 cycles and then 95°C for 1 min, 55°C for 30 s, and 95°C for 30 s. The fluorescence signals were collected between 72°C and 95°C for melting curve analysis. Cyclic threshold (Ct) value was analyzed by the 2-ΔΔCT method.
Statistical analysis
The data are presented as mean±S.D. and were statistically analyzed by one-way or two-way ANOVA for multiple-group comparison, and the calculated p values were indicated in the figures. A value of p < 0.05 was considered statistically significant.
RESULTS
PKA interacts with TDP-43
Cytoplasmic inclusions made up of hyperphosphorylated TDP-43 were found in FTLD-TDP, ALS, and AD [29–31]. To investigate whether PKA phosphorylates TDP-43, we first determined the interaction of PKA with TDP-43 by co-immunoprecipitation assay. We immunoprecipitated PKA from N2a cells with anti-PKAcα or anti-PKAcβ and analyzed the immunocomplex by western blots with anti-TDP-43 and anti-PKAcs. We found that TDP-43 was co-immunoprecipitated by PKAcα and PKAcβ (Fig. 1A), suggesting that TDP-43 may interact with PKAcα and PKAcβ.
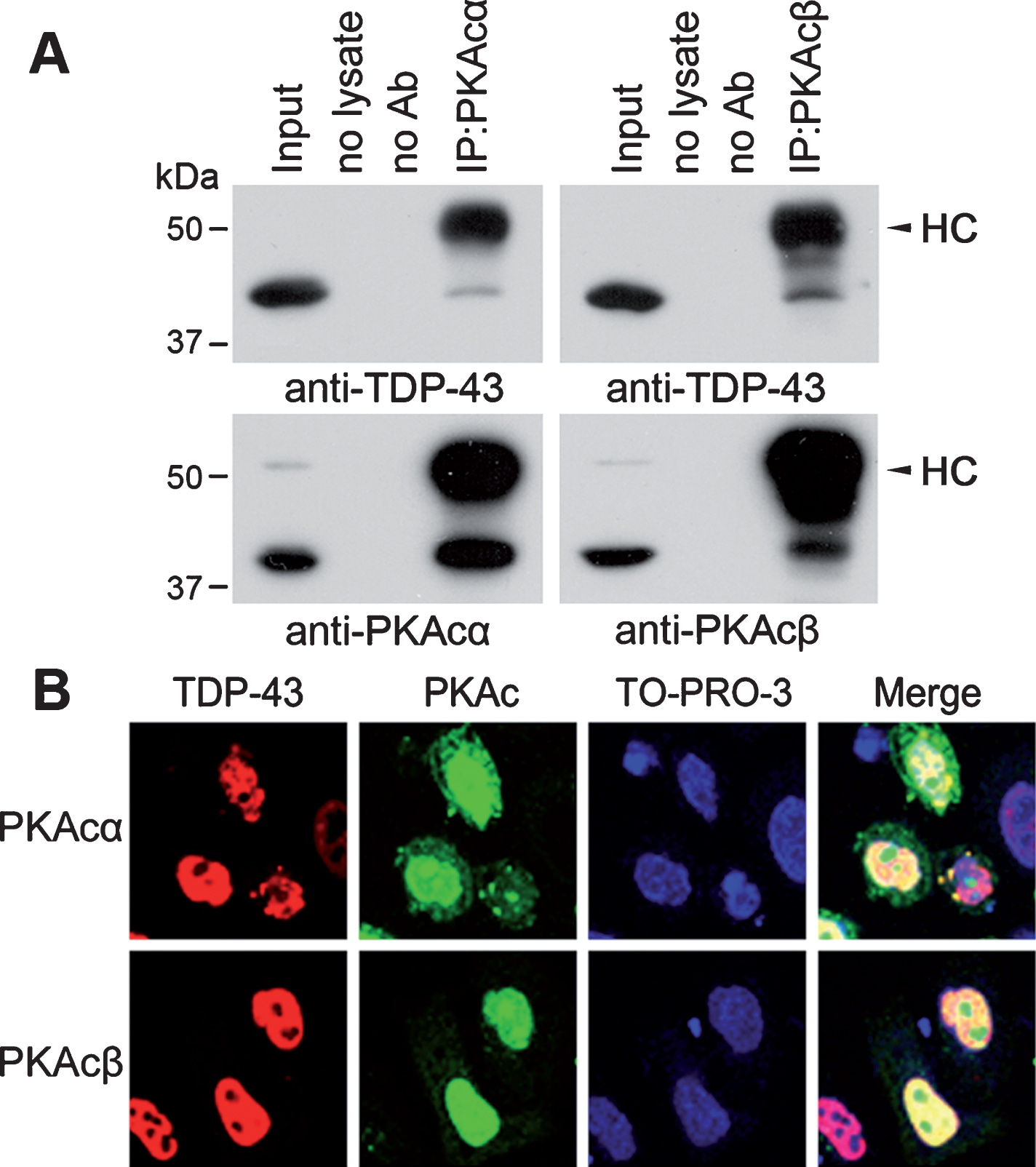
TDP-43 interacts and co-localizes in the nucleus with PKA. A) PKAcα or PKAcβ was immunoprecipitated by anti-PKAcα or anti-PKAcβ from N2a cells and analyzed by western blots with indicated antibodies. Arrowheads indicate heavy chain of IgG (HC). B) TDP-43 tagged with myc was co-expressed with PKAcα or PKAcβ tagged with HA in HeLa cells. The cells were fixed and immunostained with monoclonal anti-myc (TDP-43) or polyclonal anti-HA (PKAc) followed by Alexa 555-conjugated goat anti-mouse IgG (red) and Alexa 488-conjugated goat anti-rabbit IgG (green), respectively. TO-RPO-3 Iodide (blue) was used for nuclear staining.
To study whether PKAcs are co-localized with TDP-43 in the cells, we overexpressed TDP-43 tagged with myc with PKAcα or PKAcβ tagged with HA in HeLa cells which have easily visible cytoplasm and nucleus, immunostained the cells with monoclonal anti-myc (TDP-43) and polyclonal anti-HA (PKA). We found that TDP-43 was expressed extensively in the nucleus, while PKAcα and PKAcβ located in both nucleus and cytosol (Fig. 1B). TDP-43 was co-localized with a fraction of PKAcα or PKAcβ in the nucleus (Fig. 1B), supporting the interaction of TDP-43 with PKAcα or PKAcβ.
PKA phosphorylates TDP-43 at Ser379, Ser403/404 and Ser409/410 in vitro
To determine whether PKA phosphorylates TDP-43, we overexpressed TDP-43, PKAcα, and PKAcβ tagged with HA respectively in HEK-293FT cells and immunoprecipitated them with monoclonal anti-HA. Then we incubated IP-TDP-43 with IP-PKAcα or IP-PKAcβ at 30°C for 1 h in reaction buffer containing [γ-32P]-ATP. The reaction products were resolved by SDS-PAGE and analyzed the incorporated 32Pi by phospho-Image. We found 32P-incorporated in kinase alone and kinase with TDP-43 (Fig. 2A), suggesting autophosphorylation of PKAcs and phosphorylation of TDP-43 by PKAcα and PKAcβ, but PKAcα phosphorylated TDP-43 much more effectively than PKAcβ (Fig. 2A).
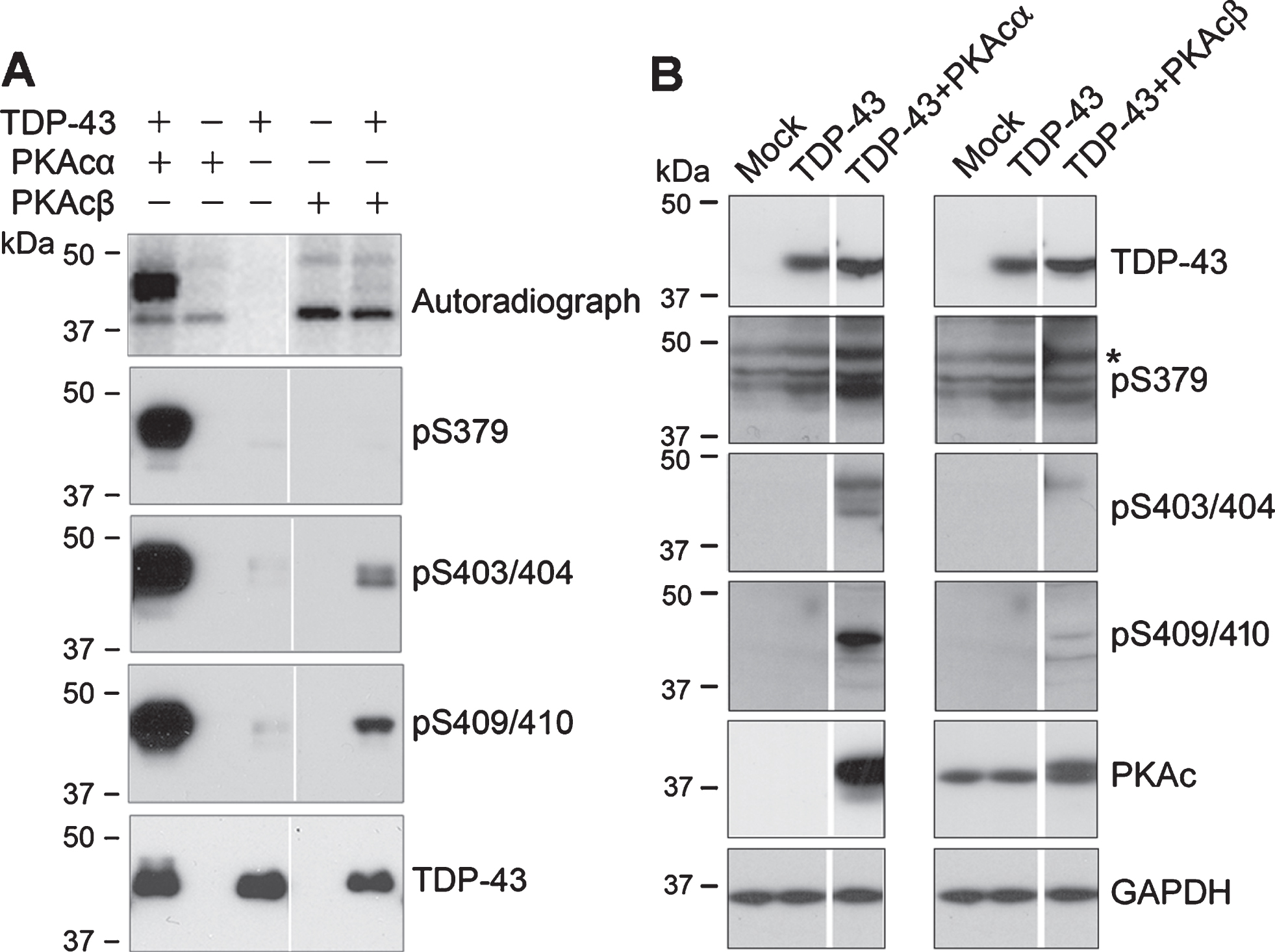
PKA phosphorylates TDP-43 at Ser379, Ser403/404, and Ser409/410 effectively. A) TDP-43, PKAcα, and PKAcβ tagged with HA were overexpressed in HEK-293FT cells and immunoprecipitated with monoclonal anti-HA respectively. IP-TDP-43 was incubated with IP-PKAcα or IP-PKAcβ in reaction buffer containing [γ-32P]-ATP or ATP at 30°C for 1 h. 32Pi incorporation of proteins was measured by autoradiography using a phospho-image device (BAS-1500, Fuji) after SDS-PAGE. Phospho-products were analyzed by western blots developed with site-specific and phosphorylation dependent anti-TDP-43. B) TDP-43 was co-transfected with PKAα or PKAcβ in HEK-293FT cells for 48 h. Site-specific phosphorylation of TDP-43 was analyzed by western blots developed with indicated antibodies. *non-specific
Parallel phosphorylation reactions were performed by replacing [r-32P]-ATP with ATP. The phospho-products were analyzed by western blots to detect the site-specific phosphorylation. We found that TDP-43 was phosphorylated in vitro at Ser379, Ser403/404, and Ser409/410 by PKAcα, and at Ser403/404 and Ser409/410 by PKAcβ (Fig. 2A). Consistently, PKAcα phosphorylates TDP-43 at these sites more efficiently than PKAcβ (Fig. 2A). Thus, we determined the phosphorylation of TDP-43 by PKAcα in the following study.
To further confirm site-specific phosphorylation of TDP-43 by PKA, we overexpressed TDP-43 together with PKAcα or PKAcβ in HEK-293FT cells and analyzed TDP-43 phosphorylation by western blots. We found that phosphorylation of TDP-43 at Ser379, Ser403/404 and Ser409/410 was clearly increased in the cells co-expressed with either PKAcα or PKAcβ (Fig. 2B), suggesting that PKA phosphorylates TDP-43 at Ser379, Ser403/404, and Ser409/40 in cultured cells.
Site-specific phosphorylation of TDP-43 by PKA is mutually enhanced
PKAcα phosphorylates TDP-43 at Ser379, Ser403/404, and Ser409/410 efficiently in vitro and in cultured cells. To confirm the site-specific phosphorylation of TDP-43 by PKA, we mutated Ser to Ala, TDP-43S379A, TDP-43S403/404A, or TDP-43S409/410A, to block the phosphorylation, overexpressed the mutants and TDP-43WT in HEK-293FT cells and immunoprecipitated them with monoclonal anti-HA. IP-TDP-43 mutants were phosphorylated with IP-PKAcα at 30°C for 1 h with [γ-32P]-ATP and analyzed by phospho-Image. We found that 32P-incorporation into TDP-43WT and TDP-43 mutants by PKAcα (Fig. 3A), confirming that PKAcα phosphorylated TDP-43. Compared with TDP-43WT, the 32P-incorporation was slightly decreased in TDP-43S379A, dramatically decreased in TDP-43S403/404A and was minimized in TDP-43S409/410A by PKAcα (Fig. 3A,B), suggesting that Ser409/410 may be the major phosphorylation site of TDP-43 by PKA or/and Ser409/410 phosphorylation may enhance TDP-43 phosphorylation by PKA.
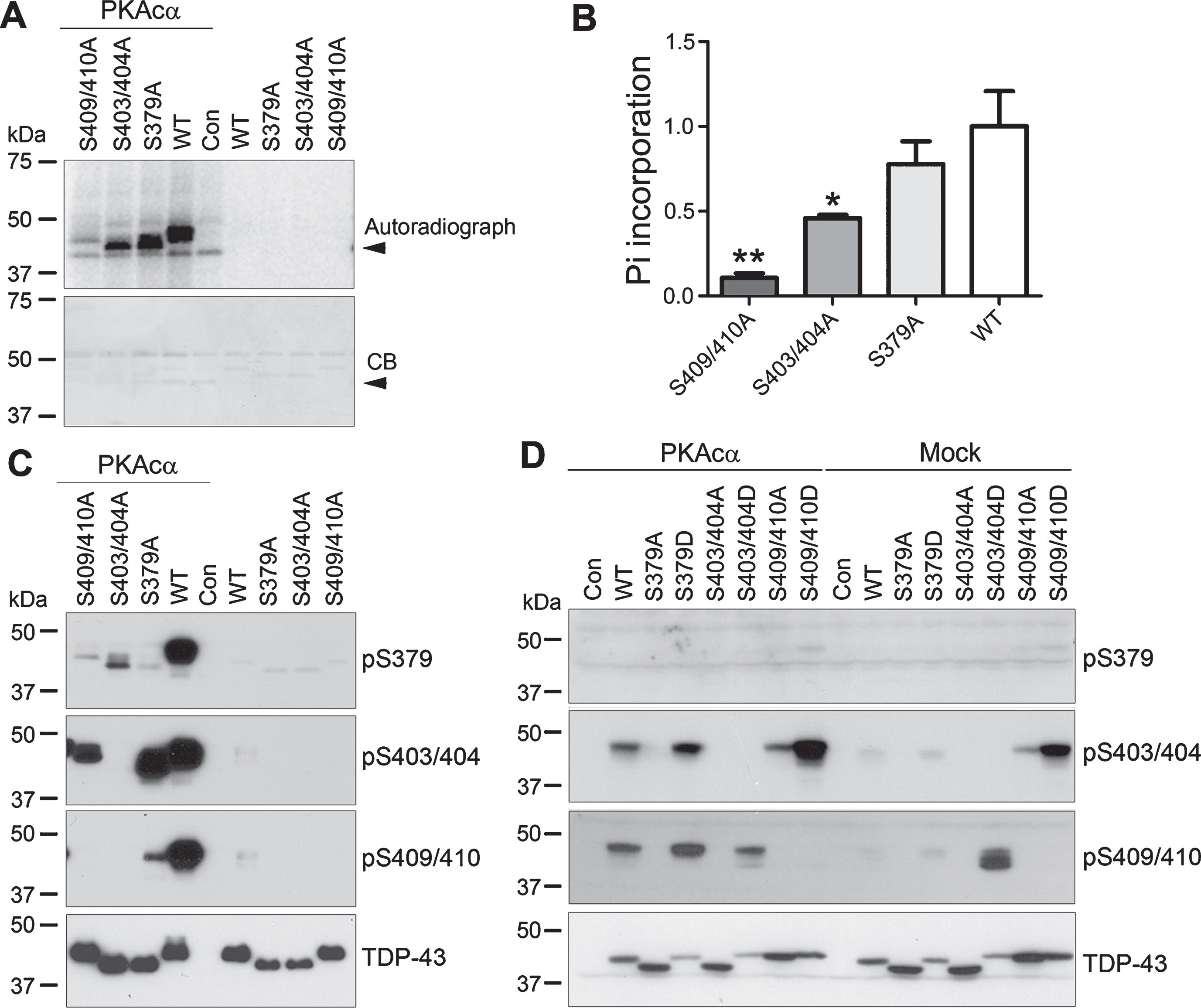
Phosphorylation of TDP-43 by PKA is enhanced site-mutually. A-C) Wild type and phospho-blocking mutants of TDP-43 were overexpressed in HEK-293FT cells and immunoprecipitated with monoclonal anti-HA and phosphorylated by IP-PKAcα in reaction buffer containing [γ-32P]-ATP (A) or containing ATP (C) at 30°C for 1 h. 32Pi incorporation of proteins was measured by autoradiography using a phospho-image device after SDS-PAGE followed by Coomassie blue staining (CB) (A) and calculated after densitometry (B). Arrowheads indicate PKA. The data were presented as mean±S.D. *p < 0.05; **p < 0.01, versus WT. C) Phosphorylation of proteins were analyzed by western blots developed with antibodies against phospho-TDP-43. D) Wild type, phospho-blocking (S379A, S403/404A, S409/410A) and phospho-mimicking (S379D, S403/404D, S409/410D) mutants of TDP-43 were expressed with/without PKAcα in HEK-293T cells for 48 h. Phosphorylation of TDP-43 was analyzed by western blots developed with indicated antibodies.
To determine the effect of phospho-blocking mutations on the site-specific phosphorylation of TDP-43, we phosphorylated TDP-43 and the phospho-blocking mutants with PKA and analyzed the phosphorylation of TDP-43 by western blots. We found that TDP-43 mutants migrated differently in western blots developed by anti-TDP-43 (Fig. 3C), suggesting that the phosphorylation may affect the conformation of TDP-43, making the mutants migrated different from the wild type TDP-43. Phosphorylation of TDP-43 at Ser379 in TDP-43S379A, at Ser403/404 in TDP-43S403/404A, and at Ser409/410 in TDP-43S409/410A was not detectable (Fig. 3C), confirming the specificity of these antibodies. Compared with TDP-43WT, TDP-43 phosphorylation by PKAcα was not changed at Ser403/404 and reduced at Ser409/410 in TDP-43S379A, was reduced at Ser379 and not detectable at Ser409/410 in TDP-43S403/404A, and was decreased at Ser379 and Ser403/404 in TDP-43S409/410A (Fig. 3C). These results suggest that phosphorylation of TDP-43 at these sites may regulate mutually its phosphorylation by PKA.
To further study the interaction of these site-phosphorylation, in addition to phospho-blocking mutations, we constructed phospho-mimicking mutations, TDP-43S379D, TDP-43S403/404D, TDP-43S409/410D, by replacing the serine residues with aspartic acid. We overexpressed the phospho-blocking and phospho-mimicking mutants with PKAcα in HEK-293T cells and analyzed the phosphorylation of TDP-43 by western blots. We found that TDP-43 at Ser379 was limited phosphorylated regardless of co-expression of PKAc (Fig. 3D). Phosphorylation of TDP-43 at Ser403/404 and Ser409/410 was not detectable or very limited in all phospho-blocking mutants, TDP-43S379A, TDP-43S403/404A, and TDP-43S409/410A, with or without PKAc co-expression (Fig. 3D), suggesting that phosphorylation of these sites may be required mutually for their phosphorylation of TDP-43 by PKA. Compared with TDP-43WT, Ser403/404 and Ser409/410 phospho-mimic mutations enhanced the phosphorylation of TDP-43 at Ser409/410 and Ser403/404, respectively, in the cells without co-expression of PKAcα (Fig. 3D). Co-expression of PKA markedly increased phosphorylation of TDP-43 at Ser403/404 and Ser409/410, as comparing with the corresponding mutants (Fig. 3D). Phospho-mimicking mutation at Ser379 enhanced the phosphorylation of TDP-43 at Ser403/404 and at Ser409/410, at Ser403/404 increased Ser409/410 phosphorylation slightly and at Ser409/410 enhanced Ser403/404 phosphorylation by PKA (Fig. 3D). Thus, these data confirm that PKA mutually phosphorylates TDP-43 at Ser379, Ser403/404, and Ser409/410.
PKA suppresses TDP-43’s function on tau mRNA processing
We recently reported that TDP-43 promoted tau mRNA instability via its 3’-UTR, leading to suppression of tau expression [18]. To study whether PKA affects this function of TDP-43, we used pEGFP/tau3′- UTR, GFP tailed with 3’-UTR of tau mRNA, to study the role of TDP-43 on tau mRNA stability by measuring GFP expression as described previously [18]. We found that overexpression of TDP-43 suppressed GFP expression at both mRNA (Fig. 4A) and protein (Fig. 4B) levels. This suppression by TDP-43 was reduced by co-expression of PKA (Fig. 4A,B), suggesting that PKA may suppress TDP-43’s function in promoting tau mRNA instability via its 3’-UTR.

PKA attenuates TDP-43-promoted tau mRNA instability. HEK-293FT cells were co-transfected with pEGFP/tau 3’-UTR and/or TDP-43, PKAcα, or PKAcβ for 48 h. The mRNA and protein levels of GFP were detected by real-time PCR (A) and western blots (B). The data were presented as mean±S.D. (n = 3). *p < 0.05, versus Con; # #p < 0.01, versus TDP-43.
We recently found that TDP-43 promotes tau exon 10 inclusion [19] and PKA phosphorylates several splicing factors and enhances tau exon 10 inclusion [26, 33]. To determine whether PKA regulates TDP-43’s function in tau exon 10 inclusion, we co-transfected tau mini-gene SI9-LI10, consisting of tau exons 9, 10, and 11 and partial intron 9 and full intron 10, as described previously [28], with TDP-43 and PKAcα, PKAcβ, or siPKAcα, siPKAcβ into HEK-293FT cells. The splicing products of tau exon 10 were then analyzed by RT-PCR. We found that TDP-43-promoted tau exon 10 inclusion was enhanced by knock-down of PKA with siRNA, but overexpression of PKA did not suppress the inclusion of tau exon 10 (Fig. 5A,B). These findings suggest that PKA might suppress TDP-43’s activity in the promotion of tau exon 10 inclusion.

PKA suppresses TDP-43-promoted tau exon 10 inclusion. A,B) TDP-43 and tau mini-gene SI9-LI10, were co-transfected with PKAcα, PKAcβ, or siRNA of PKAcα or PKAcβ (siPKAcα or siPKAcβ) into HEK-293FT cells. The splicing products of tau exon 10 were detected by RT-PCR. The ratio of inclusion/exclusion of tau exon 10 was calculated after densitometry. The data were presented as mean±S.D. ***p < 0.001, versus Con; # # #p < 0.001, versus TDP-43.
DISCUSSION
TDP-43 in cytoplasmic inclusions in FTLD-U and ALS is hyperphosphorylated at several Ser/Thr residues [4, 34–36]. In the present study, we found that PKA interacted with and phosphorylated TDP-43 at Ser379, Ser403/404, and Ser409/410. The phosphorylation of TDP-43 at these sites enhanced mutually their phosphorylation by PKA in vitro and in cultured cells. PKA overexpression ameliorated TDP-43-suppressed expression of GFP-tailed tau 3’-UTR. Knockdown of PKAcα and PKAcβ enhanced, but overexpression of PKAcs did not suppressed, TDP-43-promoted tau exon 10 inclusion. Taking together, these results suggesting PKA may phosphorylate TDP-43 and regulates its function in the regulation of mRNA, including tau mRNA processing.
None of the Ser and Thr residues in TDP-43 are followed by proline, thus non-proline directed kinases may be responsible for the phosphorylation. Several kinases, including casein kinase 1 (CK1) [4, 37], CK2 [38], CDC7 [39], and TTBK1/2 [40], are able to phosphorylate TDP-43 in vitro and in vivo and to promote its pathological accumulation and neurotoxicity [40–43]. PKA is a non-proline-directed kinase and phosphorylates many proteins. In the present study, we found for the first time that PKA also phosphorylated TDP-43 at Ser379, Ser403/404, and Ser409/410. Phospho-blocking mutations at these sites suppressed the phosphorylation of TDP-43 by PKA in vitro and in cultured cells, suggesting that prephosphorylation of TDP-43 at these sites makes it to be a better substrate for PKA and PKA phosphorylates TDP-43 at these sites mutually.
Tau is a major neuronal microtubule-associated protein. Its hyperphosphorylation and aggregation into neurofibrillary tangles are pivotal in tau pathogenesis in AD and related tauopathies [8–10, 44]. Adults human brain expresses six isoforms of tau resulted from the alternative splicing of tau exons 2, 3, and 10 [11, 12]. Dysregulation of tau exon 10 is able to cause neurodegeneration [12, 45]. No tau pathology has been seen in cerebellum of AD brain, in where ¼ level of tau is expressed, suggesting that certain expression level of tau is essential for tau pathogenesis. We recently reported that TDP-43 promotes tau mRNA instability and tau exon 10 inclusion [18, 19]. In the present study, we demonstrated that PKA phosphorylated TDP-43 and regulated its function in tau mRNA stability and tau exon 10 splicing. TDP-43 pathology was observed in up to 65% of the sporadic cases of AD [46, 47]. Thus, downregulation of PKA signaling in AD brain [26, 48] may attenuate the contribution of TDP-43 in tau pathogenesis in AD brain.
PKA also phosphorylates several splicing factors, including alternative splicing factor (ASF/SF2), SC35, 9G8, and is involved in the pre-mRNA splicing [22–24]. ASF and SC35 promoted tau exon 10 inclusion and the activity was enhanced by PKA [33, 49], while 9G8 suppressed tau exon 10 inclusion and the function was inhibited by PKA [32, 50]. Upregulation of PKA signaling enhances tau exon 10 inclusion [26]. In the present study, overexpression of PKA was not able to suppress TDP-43 promoted tau exon 10 inclusion, suggesting the role of PKA in TDP-43-promoted tau exon 10 may be ameliorated by phosphorylation of SC35 and ASF.
In summary, in the present study, we found that PKA phosphorylates TDP-43 at Ser379, Ser403/404 and Ser409/410, may suppress TDP-43-promoted tau mRNA instability and tau exon 10 inclusion, providing a plausible mechanism of PKA on TDP-43’s function linked to TDP-43 proteinopathies. Downregulation of PKA signaling in AD brain may ameliorate the contribution of TDP-43 pathology to tau pathology.