
Abstract
Background:
Neurofibrillary tangle aggregated from anomalous hyperphosphorylated tau is a hallmark of Alzheimer’s disease (AD). Trans-active response DNA-binding protein of 43 kDa (TDP-43) enhances the instability and exon (E) 10 inclusion of tau mRNA. Cytoplasmic inclusion of hyperphosphorylated TDP-43 in the neurons constitutes the third most prevalent proteinopathy of AD. Casein kinase 1δ (CK1δ) is elevated in AD brain and phosphorylates TDP-43 in vitro.
Objective:
To determine the roles of CK1δ in phosphorylation, aggregation, and function of TDP-43 in the processing of tau mRNA.
Methods:
The interaction and colocalization of TDP-43 and CK1δ were analyzed by co-immunoprecipitation and immunofluorescence staining. TDP-43 phosphorylation by CK1δ was determined in vitro and in cultured cells. RIPA-insoluble TDP-43 aggregates obtained by ultracentrifugation were analyzed by immunoblots. The instability and E10 splicing of tau mRNA were studied by using a reporter of green fluorescence protein tailed with 3’-untranslational region of tau mRNA and a mini-tau gene and analyzed by real-time quantitative PCR and reverse transcriptional PCR.
Results:
We found that CK1δ interacted and co-localized with TDP-43. TDP-43 was phosphorylated by CK1δ at Ser379, Ser403/404, and Ser409/410 in vitro and in cultured cells, which was mutually enhanced. CK1δ overexpression promoted the aggregation of TDP-43 and suppressed its activity in enhancing the instability and E10 inclusion of tau mRNA.
Conclusion:
CK1δ phosphorylates TDP-43, promotes its aggregation, and inhibits its activity in promoting the instability of tau mRNA and inclusion of tau E10. Elevated CK1δ in AD brain may contribute to TDP-43 and tau pathologies directly or indirectly.
INTRODUCTION
Transactive response DNA-binding protein of 43 kDa (TDP-43), encoded by the TARDBP gene, is a conserved RNA/DNA-binding protein ubiquitously expressing in many species [1]. As a heterogeneous nuclear ribonucleoprotein (hnRNP), TDP-43 plays important roles in gene transcription, RNA stabilization, and alternative splicing [2–4]. TDP-43 normally exists in nucleus. However, cytoplasmic inclusion of ubiquitinated and hyperphosphorylated TDP-43 aggregates is the familiar feature of frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) or amyotrophic lateral sclerosis (ALS), now termed as TDP-43 proteinopathies [5–7]. Meanwhile TDP-43 pathology constitutes the third most prevalent proteinopathy in brains of Alzheimer’s disease (AD) and also presents in other related neurodegenerative disorders including limbic-predominant age-related TDP-43 encephalopathy and chronic traumatic encephalopathy [8–11].
Neurofibrillary tangles aggregated from hyperphosphorylated tau, is considered as a hallmark of AD [12]. Through alternative splicing of the exons 2, 3, and 10, six isoforms of tau are synthesized from MAPT gene encoded mRNAs in normal human brain [13]. Exclusion or inclusion of the exon 10 produces tau isoforms containing three or four repeats of microtubule binding, termed 3R-tau or 4R-tau, respectively [14]. Adult human brain expresses proximately equally 3R-tau and 4R-tau. Disruption of this balance has been revealed in several types of tauopathies [15, 16]. We recently demonstrated that TDP-43 affected tau mRNA synthesis via promoting instability of tau mRNA and inclusion of tau exon 10 into mature mRNA [17, 18].
The function of TDP-43 is affected by phosphorylation modification [19]. And in mammals, casein kinase 1 (CK1) has about 7 isoforms (α, β, γ1, γ2, γ3, δ, ɛ) [20], among which CK1δ and CK1ɛ could phosphorylate TDP-43 in vitro [21, 22]. It was reported that CK1ɛ and CK1δ were elevated in AD brain [23, 24]. We recently found that CK1ɛ phosphorylated TDP-43, resulting in its aggregation and dysfunction, which links CK1ɛ to tau pathogenesis in AD [22, 25]. However, the roles of CK1δ in TDP-43 regulated tau mRNA processing are not very clear. We reported here that CK1δ phosphorylated TDP-43, promoted TDP-43 aggregation, and suppressed its activity in enhancing instability and exon 10 inclusion of tau mRNA. This study together with previous findings strongly suggest that elevated CK1ɛ and CK1δ, may play important roles in TDP-43 and tau pathologies in AD models.
MATERIALS AND METHODS
Plasmids, antibodies, and other reagents
CK1δ cDNA generated from RNA isolated from normal human neuronal progenitor cells by reverse transcription PCR was cloned into pCI-neo vector with hemagglutinin (HA) and Flag at the N- and C-termini, respectively and confirmed by DNA sequence analysis. The wild type, phospho-blocking (serine/Ser/S ⟶ alanine/Ala/A) or mimicking (serine/Ser/S ⟶ aspartate/Asp/D) mutants at 379, 403/404, and 409 /410 sites of TDP-43 tagged with HA in pCI-Neo, pcDNA/TDP-43·myc·his, pGEX6P1/TDP-43 and pEGFP/tau3′- UTR (GFP-fused with 3’-untranslated region/3’-UTR of tau) were obtained as addressed [17, 26]. pCI/SI9-LI10 mini-tau gene carrying tau exons 9-11, intron 10 and part of intron 9 was a gift from Dr. JH Zhou of Massachusetts University Medical School [27]. The antibodies used in this study are listed in Table 1. SiCK1δ (#sc29910) was from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Adenosine triphosphate (ATP) was purchased from Millipore Sigma (Burlington, MA, USA). [γ-32P]-ATP (#35001X01) was obtained from MP Biomedicals (Irvine, CA, USA). Protein G beads (#20399), Alexa 488 or Alexa 555 conjugated 2nd-antibodies (#A-21422, #A-11008), TO-PRO-3 iodide (642/661, #T3605), Pierce™ 660 nm protein assay kit (#22662), and enhanced chemiluminescence (ECL) Western Blotting Substrates (#32106) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Horseradish peroxidase (HRP)-conjugated 2nd antibodies (#111-035-003 and #115-035-146) were bought from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Primary antibodies used in the study
Poly-, polyclonal; Mono-, monoclonal; R, rabbit; M, mouse; p-, phosphorylated.
Cell culture and transfection
HEK-293T, HEK-293FT, and HeLa cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM) and N2a cells were cultured with DMEM/F12 supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA) at 37°C with 5% CO2. Transfections were carried out with Lipofectamine® 2000, LipofectamineTM LTX (Thermo Fisher Scientific) or FuGENE® HD (Roche Diagnostics, IN, USA) according to the instructions provided by the manufacturers.
Immunofluorescence staining
HeLa cells expressing CK1δ and TDP-43·myc·his were cultured-on glass coverslips and fixed with phosphate-buffered saline (PBS) containing 4% paraformaldehyde for 30 min at room temperature (RT). The cells were sequentially washed three times with PBS, blocked with PBS containing 10% goat serum and 0.2% Triton X-100 for 2 h at 37°C, followed with incubation with mouse anti-myc (1 : 500) and rabbit anti-CK1δ (1 : 200) overnight at 4°C. Then the cells were rinsed with PBS, incubated with 2nd antibodies conjugated with Alexa-488 or -555 (1 : 500) plus TO-PRO-3 iodide (5μg/ml) and washed with PBS. The coverslips were mounted with anti-fade mounting media (Roche Diagnostics) and photographed by a laser confocal microscope (TCS-SP2, Leica, Germany) cells in each treatment.
Immunopurification (IP) of TDP-43 and CK1δ
HEK-293FT cells expressing HA-TDP-43 or HA-CK1δ were lysed in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 50 mM NaF, 0.1% Nonidet P-40, 0.2% sodium deoxycholate, 0.1% Triton X-100, 2 mM EDTA, 1 mM Na3VO4, and a cocktail of proteinase inhibitors (1 mM AEBSF and 10μg/ml each of aprotinin, leupeptin, and pepstatin). The cell lysate was sedimented for 5 min at 15,000×g at 4°C. The protein G beads conjugated with anti-HA were incubated with cell lysate containing HA-TDP-43 or HA-CK1δ overnight at 4°C and extensively washed with lysis buffer twice and TBS twice. The immunopurified CK1δ or TDP-43 without elution were subjected to in vitro phosphorylation and western blots.
Co-immunoprecipitation (co-IP)
N2a cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM Na3VO4, 50 mM NaF, 2 mM EDTA, 1 mM AEBSF, and 10μg/ml each of aprotinin, leupeptin, and pepstatin), by probe sonication for 2 min at 20% power, 0.5 s on and 3 s off. The cell lysate was centrifuged at 15,000×g at 4°C for 5 min to yielded cell extract. The primary antibody pre-coupled protein G-beads were incubated with the extract overnight at 4°C and washed with TBS. The bound proteins were eluted and denatured by boiling in Laemmli buffer and analyzed by western blots.
Phosphorylation of TDP-43 by CK1δ in vitro
HA-CK1δ immunopurified with anti-HA from HEK-293FT cells as described above was used to phosphorylate TDP-43 in vitro. Briefly, recombinant GST-TDP-43 from E. coli or IP-TDP-43 was incubated with HA-CK1δ in reaction buffer (50 mM Tris-HCl, pH7.4, 10 mM MgCl2, 10 mM β-ME, 1 mM EGTA, and 0.2 mM [γ-32P]-ATP or ATP) at 30°C for various times. Phosphorylation products were used for analysis of autoradiography or immunoblot. For the autoradiography, phosphorylated proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained by Coomassie blue. 32Pi incorporation of proteins in the dried gel was detected by phosphorimager (Typhoon™ FLA 7000, Fuji Film, Japan).
Preparation of insoluble TDP-43 aggregates
HEK-293FT cells transiently transfected to express TDP43S409/410D were lysed in RIPA buffer (50 mM Tris-HCl, pH7.4, 150 mM NaCl, 1% NP-40, 0.5% NaDox, and 0.1% SDS) containing inhibitors of phosphatases and proteases, 50 mM NaF, 1 mM Na3VO4, 1 mM AEBSF and 10μg/ml each of leupeptin, pepstatin, and aprotinin on ice for 20 min. RIPA-insoluble and soluble fractions were obtained by centrifugation of the cell lysate for 30 min at 75,000×g at 4°C. The insoluble fraction containing aggregated TDP-43 was washed with PBS , sonicated at 80% power for 2 min, and stored at –80°C.
Western blot
Protein samples were denatured by boiling with Laemmli buffer, separated by 10% SDS-PAGE and electrically transferred onto polyvinylidene fluoride membranes (Millipore Sigma). The membrane was sequentially blocked with 5% non-fat milk dissolved in TBS for 60 min, incubated with primary antibodies in blocking buffer overnight at RT, washed three times with TBST (TBS containing 0.05% Tween), incubation with HRP-conjugated the 2nd antibodies in blocking buffer for 2 h at RT, and washed three time with TBST. The blot was incubated with ECL substrate for 1 min and exposed to HyBlot CL ® X-ray film (Denville Scientific). The film was developed and the immunosignal was quantified with Multi Gauge software V3.0 (Fuji Film).
Knockdown of CK1δ
HEK-293FT cells were transfected with short interfering RNA of CK1δ, siCK1δ (Santa Cruz) with Lipofectamine 2000. At 48 h after transfection, the cells were harvested for biochemical analyses. Transfection of scramble siRNA was used as the control.
Reverse transcription PCR (RT-PCR) and real-time quantitative PCR (qPCR)
Total RNA was prepared by using RNeasy mini kit (Qiagen, GmbH, Germany) according to the manufacturer’s instruction and used to synthesize cDNA with Omniscript reverse transcription kit (Qiagen), which was used as a template for PCR and qPCR as described below.
For measuring the alternative splicing of tau exon 10, PCR was carried out using PrimeSTARTM HS DNA Polymerase (Takara Bio Inc., Otsu, Shiga, Japan) with primers (forward, 5’-GGTGTCCACTCCCAGTTCAA-3’; and reverse, 5’-CCCTGGTTTATGATGGATGTTGCCTAATGAG-3’. The PCR products were separated by 1.5% agarose gel electrophoresis and visualized with ethidium bromide staining.
For detecting GFP expression, the qPCR was carried out with Primers (forward, 5’-TGAACCGCATCGAGCTGAAGGG-3’; reverse, 5’-ACCTTGATGCCGTTCTTCTGCTTG-3’) and FastStart Universal SYBR Green Master (ROX) (Roche Diagnostics) by using Light Cycler 96 (Roche Diagnostics). Human GAPDH (forward, 5’-CATGAGAAGTATGACAACAGCCT-3’; reverse, 5’-AGTCCTTCCACGATACCAAAGT-3’) was used as an internal control. Cyclic threshold (Ct) value was calculated by the 2-ΔΔCT method.
Statistical analysis
Data were statistically analyzed by two-way analysis of variance (ANOVA) followed by Sidak’s multiple comparisons test and by unpaired two-tailed Student’s t test for two groups comparison with GraphPad Prism 8.0 application (GraphPad Software, CA, USA). p < 0.05 was considered statistically significant.
RESULTS
CK1δ interacts with and phosphorylates TDP-43
CK1δ is elevated in AD brain [23]. Ubiquitin-positive inclusions of phosphorylated TDP-43 recently are considered as one of the pathological hallmarks of AD [8, 28]. To explore the role of CK1δ in TDP-43 pathogenesis, we firstly determined the interaction of CK1δ with TDP-43. We immunoprecipitated TDP-43 from N2a cell lysate with monoclonal anti-TDP-43 (H8) and analyzed the immuno-complexes with western blots developed with polyclonal anti-TDP-43 (A260) or anti-CK1δ. We found that CK1δ was co-precipitated by anti-TDP-43 (Fig. 1A), indicating a possible interaction between CK1δ and TDP-43.

TDP-43 interacts with and is phosphorylated by CK1δ. A) TDP-43 was immunoprecipitated with monoclonal anti-TDP-43 (H8) from N2a cell lysate. The immuno-complex was analyzed by western blots developed with anti-CK1δ and anti-TDP-43 (A260). B) pCI/HA·CK1δ·Flag was co-transfected with pcDNA/TDP-43·myc·his into HeLa cells. The cells were immunofluorescence-stained with anti-CK1δ (red) and anti-myc (for TDP-43, green). Nucleus was stained by TO-RPO-3 iodide (blue). Scale bar, 25μm. C) Recombinant GST-TDP-43 was phosphorylated by immuopurified-CK1δ for 0–2 h. The phosphorylation products were analyzed by western blots developed with indicated antibodies. Arrowhead, TDP-43.
We then analyzed the localization of CK1δ and TDP-43 in cultured HeLa cells. We co-expressed TDP-43·myc and CK1δ in HeLa cells, immunofluorescence-stained the cells with polyclonal anti-CK1δ and monoclonal anti-myc (for TDP-43) and visualized with confocal microscope. We observed that CK1δ was presented in both cytoplasm and nucleus, but TDP-43 mainly located in the nucleus (Fig. 1B), suggesting the partial co-localization of CK1δ and TDP-43.
To determine the phosphorylation of TDP-43 by CK1δ, immunopurified (IP)-CK1δ from HEK-293FT cells was incubated with recombinant GST-TDP-43 in the phosphorylation reaction buffer for 0–2 h, then the reaction products were subjected to western blots to analyze TDP-43 phosphorylation. We found phosphorylated TDP-43 at Ser379, Ser403/404, and Ser409/410 by the incubation with CK1δ, which was increased in the incubation time-dependent manner (Fig. 1C). Thus, these results suggest that CK1δ phosphorylates TDP-43 at above sites in vitro.
Site-specific phosphorylation of TDP-43 by CK1δ is mutually enhanced
Above study showed that TDP-43 was phosphorylated by CK1δ at Ser379, Ser403/404, and Ser409/410. To confirm the site-specific phosphorylation, we mutated the serine (Ser, S) residues of TDP-43 at 379, 403/404, 409/410 to alanine (Ala, A) to block the phosphorylation at these sites. Then we overexpressed wild type of TDP-43 (TDP-43WT) and above phospho-blocking mutants tagged with HA individually in HEK-293FT cells and immunopurified them with anti-HA. The IP-TDP-43 s were phosphorylated with the IP-CK1δ in the reaction buffer containing [γ-32P]-ATP for 1 h at 30°C. The reaction products were resolved by 10% SDS-PAGE followed by Coomassie blue (CB) staining. 32Pi incorporation was assessed by autoradiography. We observed an obvious phospho-signal at 50 kDa in CK1δ alone control (Fig. 2A), indicating that IgG heavy chain and/or CK1δ were phosphorylated by CK1δ. There was a stronger 32Pi signal in CK1δ with TDP-43WT, confirming the phosphorylation of TDP-43 by CK1δ. Of note, compared with TDP-43WT, the incorporation of 32Pi was decreased in all the TDP-43 phospho-blocking mutants catalyzed by CK1δ, supporting that 379, 403/404 and 409/410 sites are phosphorylated by CK1δ.
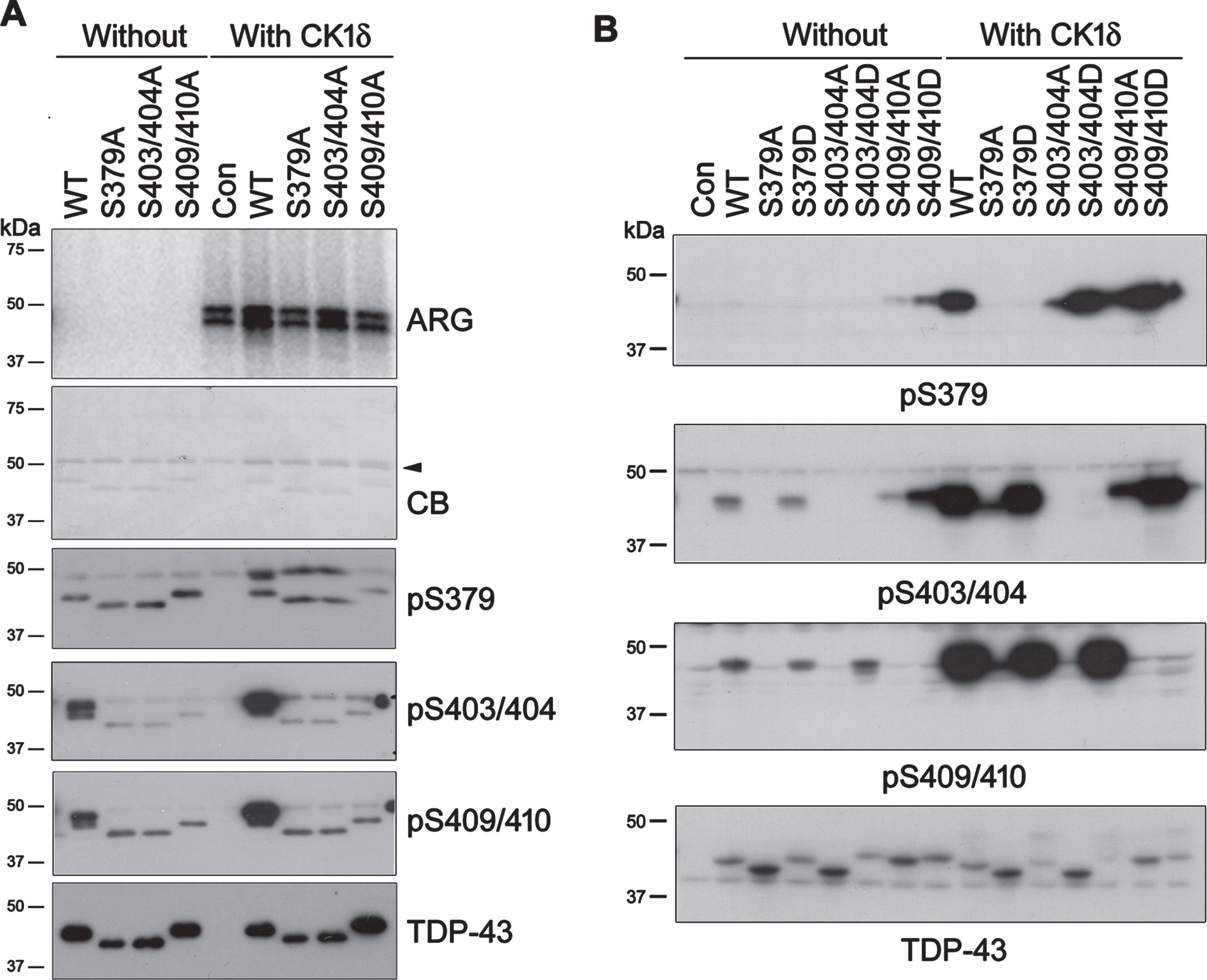
Phosphorylation of TDP-43 by CK1δ at Ser379, Ser403/404, and Ser409/410 is enhanced mutually. A) HA-tagged TDP-43, its phospho-blocking mutants and CK1δ were overexpressed separately in HEK-293FT cells, and immunopurified (IP) with anti-HA respectively. IP-TDP-43 s were phosphorylated by IP-CK1δ in reaction buffer containing [γ-32P]-ATP at 30°C for 1 h. Phospho-products was resolved by SDS-PAGE followed by Coomassie blue (CB) staining. 32Pi incorporation was assessed by autoradiography (ARG). The phosphorylation reaction was carried out parallelly with ATP. The reaction products were determined by western blots. Arrowhead, heavy chain of IgG. B) TDP-43, its phospho-blocking/mimicking mutants were overexpressed in HEK-293T cells together with CK1δ. Phosphorylated TDP-43 was analyzed by western blots developed with indicated antibodies.
We analyzed the site-specific phosphorylation of the phospho-blocking mutants of TDP-43 by CK1δ in parallel experiment with western blots (Fig. 2A). Similar as we found before [25, 26], TDP-43 mutants showed different apparent molecular weight in TDP-43 blot (Fig. 2A). Western blots also showed that Ser379, Ser403/404, and Ser409/410 of TDP-43 were phosphorylated by CK1δ (Fig. 2A). Compared with TDP-43WT, TDP-43 phosphorylation by CK1δ was decreased at Ser379 in TDP-43S403/404A and TDP-43S409/410A, and was almost undetectable at Ser403/404 in TDP-43S379A and TDP-43S409/410A, at Ser409/410 in TDP-43S379A and TDP-43S403/404A (Fig. 2A). Thus, each of these phospho-blocking mutations suppressed the phosphorylation of TDP-43 by CK1δ at other sites. Ser379, Ser403/404 and Ser409/410 phosphorylation may mutually regulate TDP-43 phosphorylation by CK1δ in vitro.
To further explore the site-specific phosphorylation of TDP-43 by CK1δ in cultured cells, in addition to phospho-blocking mutations as described above, we mutated Ser residues at these sites to aspartate (Asp, D) to mimic phosphorylation. The phospho-blocking and phospho-mimicking mutants were expressed in HEK-293T cells together with CK1δ. Their phosphorylation was analyzed by western blots, showing that phosphorylated TDP-43 at Ser379, Ser403/404, and Ser409/410 was almost undetectable in all phospho-blocking mutants (Fig. 2B), supporting that these sites phosphorylation by CK1δ is required mutually. Oppositely, we found that compared with TDP-43WT, phospho-mimicking mutation of Ser379 increased Ser403/404 and Ser409/410 phosphorylation; Ser403/404-phospho-mimicking mutation enhanced Ser379 and Ser409/410 phosphorylation; and Ser409/410-phospho-mimicking mutation increased Ser379 and Ser403/404 phosphorylation in the cells co-expressed with CK1δ (Fig. 2B). These results further support that TDP-43 phosphorylation at Ser379, Ser403/404 and Ser409/410 by CK1δ is enhanced mutually.
CK1δ promotes TDP-43 aggregation
We previously found that phospho-mimicking mutants, especially TDP-43S409/410D, was easy to aggregate [25]. Amyloid-type aggregates of protein are detergent-insoluble. To compare the phosphorylation of aggregated TDP-43 with its soluble form, we expressed HA-TDP-43S409/410D in HEK-293FT cells and lysed the cells with RIPA buffer. RIPA-insoluble and -soluble fractions were obtained by ultracentrifugation and analyzed by western blot analyses. We observed that RIPA-soluble TDP-43 displayed one band, but RIPA-insoluble TDP-43 showed more than one band in the anti-HA blot (Fig. 3A). Compared with RIPA-soluble TDP-43, RIPA-insoluble TDP-43 was phosphorylated at Ser379 and Ser403/404. Of note, SDS- and reducing regent-resistant high molecular weight TDP-43 (HMW-TDP-43) was observed in the RIPA-insoluble fractions by long time exposure (Fig. 3A). These data suggest that phosphorylated and HMW-TDP-43 is enriched in RIPA-insoluble fraction, which contains insoluble TDP-43 aggregates.
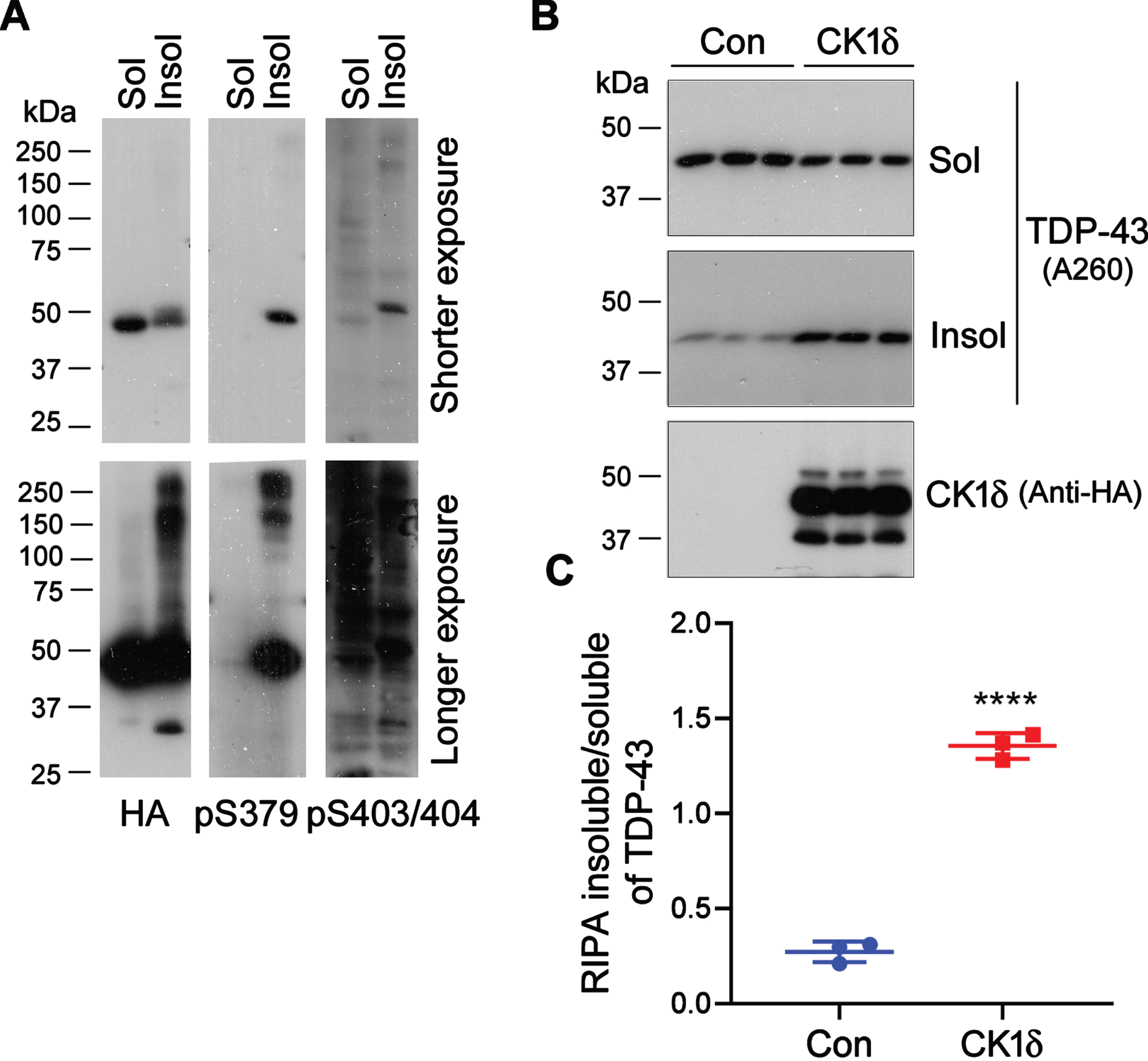
CK1δ promotes the aggregation of TDP-43. A) HA-TDP-43S409/410D was expressed in HEK-293FT cells. The cells were lysed in RIPA-buffer. RIPA-insoluble and -soluble fractions were analyzed by western blots developed with indicated antibodies. B) HEK-293FT cells expressing HA-CK1δ were lysed with RIPA buffer. RIPA-insoluble and -soluble TDP-43s were analyzed. The ratios of RIPA insoluble/soluble TDP-43 are presented as scatter plots with mean±S.D. (n = 3). ****p < 0.0001.
To investigate the effect of CK1δ on TDP-43 aggregation, we overexpressed CK1δ in HEK-293FT cells and determined RIPA-insoluble and -soluble TDP-43 s by immunoblots. We found that RIPA-insoluble TDP-43 levels were significantly increased, and the RIPA-soluble TDP-43 levels were slightly decreased in the cells with CK1δ overexpression (Fig. 3B), leading to a significant increase of the ratio of RIPA insoluble/soluble TDP-43 (Fig. 3C). Thus, CK1δ overexpression may promote the aggregation of TDP-43.
CK1δ suppresses TDP-43’s ability in enhancing tau mRNA instability
We reported recently that TDP-43 suppressed tau expression by enhancing tau mRNA instability via its 3’-UTR [17]. To determine the effect of CK1δ on TDP-43 regulated tau mRNA stability, 3’-UTR of tau mRNA tailed GFP (Fig. 4A) together with TDP-43 and CK1δ were transfected into HEK-293FT cells. The expression of GFP was analyzed by qPCR and western blots. We found consistently that TDP-43 overexpression inhibited the expression of GFP at both mRNA and protein levels (Fig. 4B, C). The TDP-43 suppressed GFP-expression was reversed by co-expression of CK1δ (Fig. 4B, C). Taking the advantage of the green fluorescence of GFP, we found a decrease of fluorescence signaling in the cells with TDP-43 overexpression, which was attenuated by co-overexpression of CK1δ (Fig. 4D). These results suggest that CK1δ suppresses TDP-43’s ability in enhancing the instability of tau mRNA through its 3’-UTR.

CK1δ suppresses TDP-43’s function in enhancing tau mRNA instability. A) Schematic of GFP tailed with tau mRNA 3’-UTR. B) HEK-293FT cells were expressed with tau 3’-UTR together with TDP-43 and CK1δ. The mRNA level of GFP was analyzed by qPCR and presented as mean±S.D. (n = 4). ***, # # #p < 0.001; *versus Con; #versus TDP-43. C) GFP protein in cell lysate was analyzed by western blots. D) Representative green fluorescence of cells after co-expression of GFP with TDP-43 or/and CK1δ for 48 h.
CK1δ suppresses TDP-43’s function in enhancing tau exon 10 inclusion
To learn the effect of CK1δ on TDP-43 enhanced inclusion of tau exon 10, we transfected pCI/SI9-LI10 mini-tau gene (consisting of tau exons 9-11, partial intron 9 and full intron 10) (Fig. 5A) together with TDP-43, CK1δ, or siCK1δ into HEK-293FT cells and analyzed the splicing products of tau exon 10 by RT-PCR. The results showed that overexpression TDP-43 increased the ratio of inclusion/exclusion of tau exon 10, which was attenuated by CK1δ overexpression (Fig. 5B) and enhanced by knock-down of CK1δ with siCK1δ (Fig. 5C). These data suggest that CK1δ suppresses TDP-43’s function in enhancing the inclusion of tau exon 10.

CK1δ suppresses TDP-43-enhanced tau exon 10 inclusion. A) Schematic of tau mini-gene SI9-LI10. E, exon; I, intron. B,C) HEK-293FT cells were co-transfected with pCI/SI9-LI10, TDP-43 and CK1δ (B), or siCK1δ (C). Tau exon 10 splicing products were analyzed by RT-PCR. The ratios of inclusion/exclusion of tau exon 10 are presented as mean±S.D. (n = 4). #p < 0.05; ****, # # # #p < 0.0001; *versus Con; #versus TDP-43.
DISCUSSION
TDP-43 involves in modulating tau mRNA production by affecting instability and inclusion of tau exon 10 into tau mRNA [17, 18]. CK1δ is increased in AD brains [29]. Herein we reported that CK1δ interacted with TDP-43, phosphorylated TDP-43 at Ser379, Ser403/404 and Ser409/410 sites, and the site-specific phosphorylation was enhanced mutually. Overexpression of CK1δ promoted TDP-43 aggregation and induced TDP-43 dysfunction, which lead to enhancing instability of tau mRNA and inclusion of tau exon 10. These results together with previous findings stand that elevated CK1δ in AD brains may involve to TDP-43 pathology and tau pathology via suppressing its activity in modulation of tau mRNA processing. CK1δ could be a therapeutic target for AD.
Aggregated TDP-43 in inclusion is hyperphosphorylated at many sites in affected neurons [5, 30]. A few kinases, such as CK1 and CK2 [5, 31], tau tubulin kinases 1 and 2 [32–34], cell division cycle 7 [35], and cAMP-dependent protein kinase [26], have been revealed to phosphorylate TDP-43. CK1ɛ and CK1δ belong to CK1 family of Ser/Thr protein kinases. Their mRNA levels are upregulated in AD brains [36]. Compared to controls, CK1ɛ and CK1δ increased greater than 30-fold in AD hippocampus [29]. TDP-43 at Ser379, Ser403/404 and Ser409/410 sites could be phosphorylated by CK1 in vitro [5]. We recently reported that CK1ɛ phosphorylated TDP-43, promoted its aggregation and suppressed its function [22, 25]. Here, we found that, being similar to CK1ɛ, CK1δ also phosphorylated Ser379, Ser403/404 and Ser409/410 groups of TDP-43 and promoted its aggregation. Phospho-blocking mutations (TDP-43S409/410A, TDP-43S403/404A, or TDP-43S379A) almost completely blocked TDP-43 phosphorylation at other sites by CK1δ, while the phospho-mimicking mutations (TDP-43S379D, TDP-43S403/404D, or TDP-43S409/410D) enhanced the phosphorylation of TDP-43 at other sites. Thus these results support that the phosphorylation of TDP-43 in site-specific style by CK1δ was enhanced mutually.
TDP-43 is predominantly expressed in the nucleus; and is phosphorylated then transferred to cytoplasm forming inclusion in neuron of individuals with ALS and FTLD-TDP [6, 7]. Phosphorylation modification of TDP-43, catalyzed by CK1δ or CK1ɛ, led to its mislocalization and accumulation in cytoplasm from nucleus [21, 22]. Phospho-mimicking mutation at Ser379, Ser403/404 and Ser409/410 sites increases its aggregation in cytoplasm [25]. In this study we found that RIPA-insoluble TDP-43 was phosphorylated and displayed SDS- and reducing reagent resistant-HMW-TDP-43 species. Forced expression of CK1δ boosted aggregation of TDP-43. Considering CK1δ level is dramatically increased in AD brains [29], we speculate that elevated CK1δ together with CK1ɛ may contribute the TDP-43 pathogenesis progress in AD brain.
NFTs are made up of anomalous hyperphosphorylated tau in the brain of individuals with AD and related tauophthies [37, 38]. CK1δ is a non-proline directed kinase and phosphorylated tau at several sites, suggesting that elevated CK1δ may affect tau pathogenesis of AD through phosphorylating tau protein [39, 40].
The development of tau pathology orderly follows a stereotypical pattern from entorhinal cortex, limbic system, and isocortex region in AD brains [41, 42]. Tau pathology appeared only in transgenic rodents with overexpression of wild type or mutated human tau [16, 43– 46], but it has not been observed in non-transgenic normal mice [47]. Rat cerebellum only expresses about as 1/4 level of tau as does in cerebral cortices [48], AD cerebellum lacks tau pathology [49], and decreased tau expression protects AD mice against cognitive impairment [50]. These findings suggest that tau level may be crucial for progress of tau pathology. We recently discovered that TDP-43 inhibited tau expression by acting on the 3’-UTR to promote its mRNA instability [17]. Here, by using a reporter of tau mRNA 3’-UTR tailed GFP, we demonstrated that ectopic expression of CK1δ rescued TDP-43 suppressed GFP expression, suggesting that phosphorylation or/and aggregation of TDP-43 by CK1δ may inhibit the activity of TDP-43 in decreasing tau mRNA stability.
In normal adult human brains, 3R-tau and 4R-tau produced from alternative splicing of tau exon 10 are approximately equally expressed [51]. Dysregulation of tau exon 10 splicing can lead to several types of tauopathies [52]. We previously found that TDP-43 enhanced tau exon 10 inclusion [18]. Here we showed that ectopic expression of CK1δ suppressed, while knockdown of CK1δ promoted TDP-43-enhanced tau exon 10 inclusion, suggesting that CK1δ suppressed the activity of TDP-43 in modulation of tau exon 10 splicing. Taking together, we propose that in addition to direct phosphorylation of tau as reported previously [24], elevated CK1δ in AD brains may accelerate tau pathology by suppressing the function of TDP-43 in enhancing the instability of tau mRNA and the inclusion of tau exon10.
Cytoplasmic aggregates of hyperphosphorylated TDP-43 may induce disease through a toxic functional loss, dysregulating mRNA processing implicated in neuronal function [5, 54]. However, whether phosphorylation by CK1δ suppresses TDP-43’s function directly or by promoting the cytoplasmic aggregation remain to be determined.
In summary, we find here that TDP-43 is phosphorylated by CK1δ at Ser379, Ser403/404 and Ser409/410. Overexpression of CK1δ, boosts TDP-43 aggregation and restrains its activity on enhancing the instability and exon 10 inclusion of tau mRNA. Elevated CK1δ expression in AD brains may not only contribute to TDP-43 pathogenesis, but also accelerate tau pathology by phosphorylating tau directly and by promoting tau expression and dysregulating tau exon 10 splicing through TDP-43 (Fig. 6).
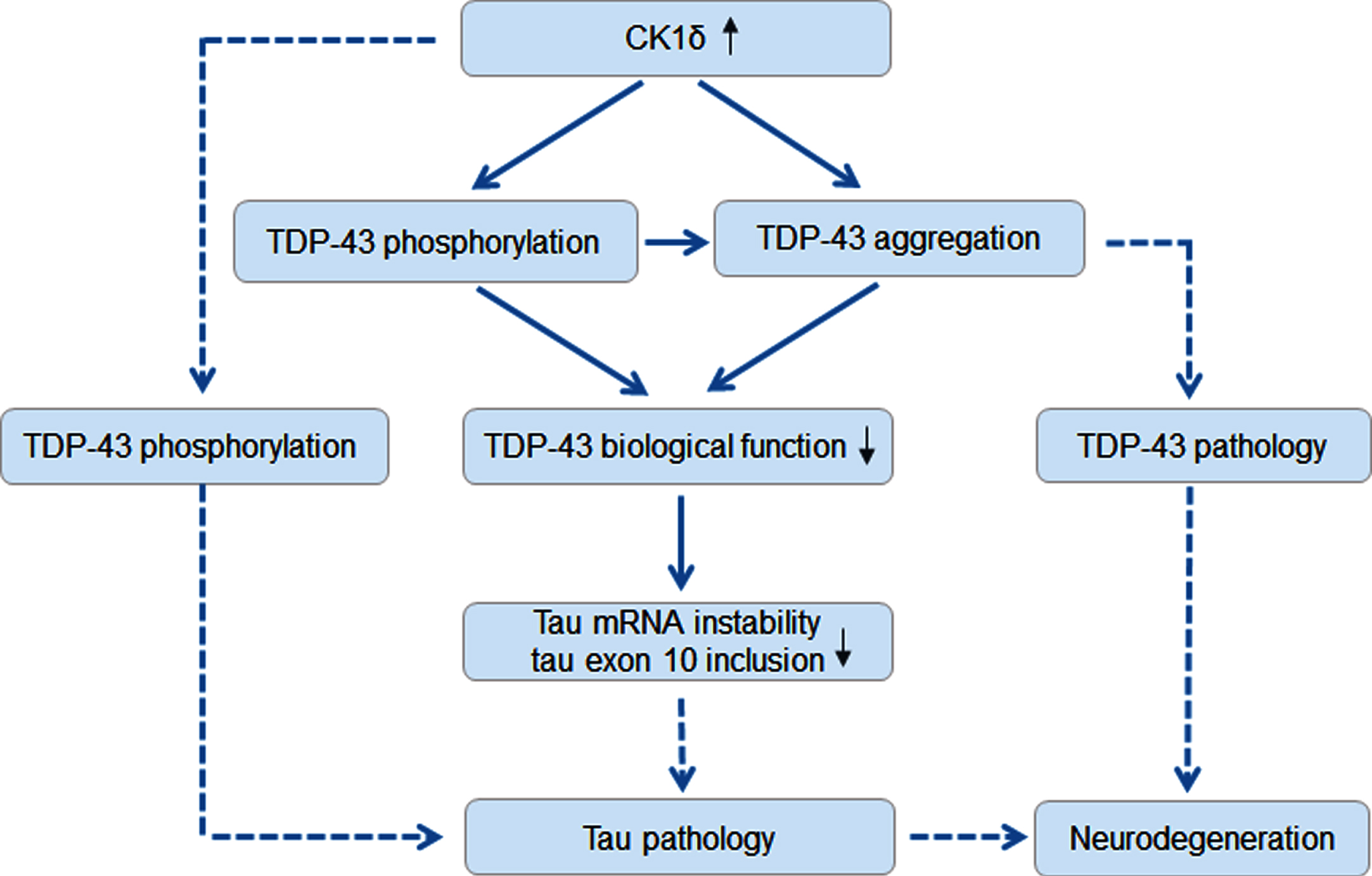
Proposed molecular mechanism of CK1δ involved in TDP-43 and tau pathologies in AD brain. Solid line presents the results from the present study and dash line presents previous findings.
Footnotes
ACKNOWLEDGMENTS
We are thankful to Dr. Jianhua Zhou for his generous gift of tau mini-gene plasmid pCI/SI9-LI10.
FUNDING
This study was supported by Natural Science Foundation of Jiangsu Province (BK20211329), National Natural Science Foundation of China (32171258, 31870772, 31671046), National Projects of University Students’ Innovation and Entrepreneurship Program (202210304038Z), U.S. Alzheimer’s Association grant DSAD-15-363172, and by funds from the New York State Office for People With Developmental Disabilities and Co-innovation Center of Neuroregeneration of Nantong University.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author.