
Abstract
Background:
Axonal injury has been implicated in the development of amyloid-β in experimental brain injuries and clinical cases. The anatomy of the spinal cord provides a tractable model for examining the effects of trauma on amyloid deposition.
Objective:
Our goal was to examine the effects of axonal injury on plaque formation and clearance using wild type and 5xFAD transgenic Alzheimer’s disease mice.
Methods:
We contused the spinal cord at the T12 spinal level at 10 weeks, an age at which no amyloid plaques spontaneously accumulate in 5xFAD mice. We then explored plaque clearance by impacting spinal cords in 27-week-old 5xFAD mice where amyloid deposition is already well established. We also examined the cellular expression of one of the most prominent amyloid-β degradation enzymes, neprilysin, at the lesion site.
Results:
No plaques were found in wild type animals at any time points examined. Injury in 5xFAD prevented plaque deposition rostral and caudal to the lesion when the cords were examined at 2 and 4 months after the impact, whereas age-matched naïve 5xFAD mice showed extensive amyloid plaque deposition. A massive reduction in the number of plaques around the lesion was found as early as 7 days after the impact, preceded by neprilysin upregulation in astrocytes at 3 days after injury. At 7 days after injury, the majority of amyloid was found inside microglia/macrophages.
Conclusion:
These observations suggest that the efficient amyloid clearance after injury in the cord may be driven by the orchestrated efforts of astroglial and immune cells.
INTRODUCTION
Amyloid-β protein precursor (AβPP) is constantly synthesized and transported anterogradely along axons to the nerve terminals for normal synaptic function [1–3]. Upon injury to axons, transport is affected and AβPP accumulates focally where, by still unknown mechanisms, it is aberrantly cleaved to form amyloid-β [4]. It has been observed in traumatic brain injury (TBI) patients that amyloid-β plaques appear shortly after injury, regardless of the patient’s age [5, 6]. It has also been shown that surviving TBI patients exhibit amyloid-β pathology in their brains chronically and they are more likely to develop neurodegenerative diseases such as Alzheimer’s disease (AD) later in life [7–9]. The mechanistic link between TBI and amyloid-related neuropathology is not entirely clear. MRI and PET scanning using 11C-Pittsburgh compound B (11C-PiB), a radioactive thioflavin T analog that binds amyloid-β plaque [10], revealed a different pattern of tracer binding in AD and TBI patients, suggesting a different mechanism of amyloid deposition. Moreover, observations that 11C-PiB binding in cortical regions together with white matter damage in connected tracts suggested that aggregation of amyloid plaque after TBI is due to axonal injury [9].
Animal models were developed to investigate the link between TBI and deposition of amyloid-β. It has been shown that amyloid-β accumulates in axonal bulbs and brain parenchyma using head rotational acceleration injury in pigs [11]. Similar accumulation of amyloid-β in axonal bulbs can be seen in rabbits receiving the same injury and Sprague-Dawley rats receiving impact head injury [12, 13]. More experimental studies were conducted with transgenic mice due to the fact that wild type (WT) mice do not normally accumulate amyloid plaque after injury [14]. In transgenic mice overexpressing human APP and/or presenilin gene mutations, most injury studies found an acute increase of amyloid-β peptide as measured by western blot or ELISA [15, 16]. However, conflicting results in the ability of these transgenic mice to promote amyloid plaque formation after injury are reported. For example, controlled cortical impact in 3xTg mice induces diffuse plaque formation and intra-axonal accumulation of amyloid-β [16, 17]. In contrast, the same injury in PDAPP mice, which express mutant APP driven by a platelet derived growth factor promoter, reduces plaque load [18, 19]. Yet other studies showed no shift in plaque dynamics after TBI in PDAPP mice or APP/PS1 mice [20, 21]. Taken together, while there appears to be a stimulation of amyloid-β deposition by trauma in both humans and animals, the relationship is not always direct, and more work is required to explain the reported discrepancies, and their significance.
We previously mapped the amyloid-β plaque load in the 5xFAD mouse spinal cord and identified novel amyloid-β-positive thread-like structures in the peri-axonal space of spinal axons [22]. The 5xFAD mouse carries five human familial AD gene mutations, with three in the gene encoding amyloid precursor protein (APP) and the other two in the presenilin-1 gene [23]. These animals develop extensive and more rapid amyloid pathology, showing plaque deposition as early as 2 months in the brain, compared to other AD models [24]. Given the robust relationship between brain trauma and amyloid accumulation, in this report we used spinal cord injury (SCI) as an alternative to TBI to examine the effects of axonal injury on plaque formation and clearance, in a region of the central nervous system not usually prone to plaque deposition. The longitudinal anatomy of spinal tracts, together with the ability to deliver a focal traumatic injury, enabled us to examine the spatial relationship between the locus of injury and amyloid-related pathological changes above and below the lesion. We used contusion injury at low thoracic cord which additionally allows functional assessment to evaluate the effect of impact on the animals. Unexpectedly, rather than promoting plaque accumulation, our results showed that, when the injury was induced before plaques were spontaneously deposited, SCI prevented accumulation of plaque at considerable distances from the injury site. More intriguingly, focal mechanical injury promoted rapid clearance of pre-existing plaques, possibly with the help of astrocytes and immune cells.
MATERIALS AND METHODS
Animals
All animal experiments were approved by the Animal Care Committee at the University of Calgary. 5xFAD mice (Tg6799) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA), the colony was maintained by crossing heterozygous transgenic mice with WT mice. Genotyping was performed by polymerase chain reaction analysis of ear sample DNA. WT littermates were used as controls. A total of 61 mice were used in the study.
Spinal cord injury and post-operative care
Mice were deeply anaesthetized with 1.5% isoflurane with oxygen. The area was shaved and sterilized with isopropanol and betadine. Buprenorphine (0.05 mg/kg) and Baytril (5 mg/kg) were given before surgery. A midline incision was made to expose the vertebral column, and laminectomy was then performed at the tenth thoracic vertebra, corresponding to the twelfth thoracic (T12) spinal level. The vertebrae above and below the exposed spinal cord were stabilized with a pair of clamping forceps. The cord was then placed under the impact tip of an Infinite Horizon spinal cord impactor. An impacting force of 75 kdynes was used. The muscles and skin were sutured in layers and the animals were returned to their home cage for recovery on a heating pad. Post-operative care included buprenorphine and Baytril administration for 2 days after surgery. Manual voiding of bladders was performed twice daily until return of bladder function.
For unilateral contusion at cervical level (n = 5), 6-week-old 5xFAD mice were used. We exposed the cervical vertebral column, and laminectomy was then performed at the sixth cervical vertebra. To avoid excessive damage to the cord, we placed stretched parafilm on top of the exposed cord and an impact tip of 0.6 mm diameter was used to impact the spinal cord at a force at 65 kdynes. Wound closure after surgery and post-operative care were as described above. Mice were kept for 8 weeks before tissue harvest.
For hemisection at the thoracic level (n = 3), 27-week-old 5xFAD mice were used. We exposed the thoracic vertebral column and laminectomy was performed at the tenth thoracic vertebra. We used a pair of spring scissors to carefully excise the dorsal right side of the spinal cord. Mice were kept for 7 days before tissue harvest.
Behavioral assessment
Mice were screened for pre-existing abnormality and allowed to become accustomed to the testing environment before surgery. After low thoracic contusion surgery, hindlimb function was monitored according to the Basso Mouse Scale for locomotion (BMS) [25]. Briefly, the mouse was placed in a 2 m diameter circular plexiglass tank. It was allowed to move freely in the tank for 4 min and its locomotor performance was scored by two blinded observers according to the scoring sheet provided in their publication. Scores for left and right sides were averaged for each animal.
Perfusion and tissue processing
Animals were given a lethal dose of sodium pentobarbital and perfused intracardially with normal saline followed by ∼50 ml of fixative containing 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The brain and spinal cord were harvested, post-fixed with fresh fixative overnight, and subsequently placed in 30% phosphate buffered-sucrose. After the samples had sunk, the cords were embedded in optimum cutting temperature compound (VWR, Suwanee, GA, USA), frozen in pre-cooled isopentane, and cut in cross or sagittal sections at a thickness of 10μm on a cryostat. The sections were collected on SuperFrost Plus slides (VWR, Chester, PA, USA) and stored at –20°C for later use.
Immunohistochemistry
For immunolabeling involving amyloid-β, antigen retrieval with formic acid was performed unless otherwise noted. Briefly, spinal cross or sagittal sections were washed with 0.01 M phosphate buffered saline (PBS, pH 7.4) and treated with 98% formic acid for 5 min at room temperature. After washing with PBS, the sections were incubated in blocking solution containing 10% normal serum, 1% bovine serum albumin, and 0.3% triton X-100 for 1 h at room temperature. The sections were subsequently incubated in primary antibodies at 4°C for 14–16 h. Primary antibodies included monoclonal anti-amyloid-β (1–16), clone 6E10 (1:1000, Biolegend, San Diego, CA, USA); polyclonal anti-amyloid-β 1–42 (Aβ42, 1:200, Cat # AB5078P, Millipore, Temecula, CA, USA); rabbit anti-glial fibrillary acidic protein (GFAP, 1:500, Cat # Z0334, Dako, Glostrup, Denmark); rabbit anti-ionized calcium-binding adapter molecule 1 (Iba-1, 1:200, Cat # 019–19741, Wako, Osaka, Japan); and goat anti-neprilysin. (1:50, Cat # AF1126, R & D Systems, Minneapolis, MN, USA). Primary antibodies were omitted in secondary-only controls. After 3x washes (15 min each) in PBS-tween 20 (0.05%, PBS-T), the sections were incubated in the corresponding fluorescent conjugated secondary antibodies (1:500, Invitrogen, Eugene, OR, USA) in PBS for 1 h at room temperature. After three washes in PBS-T, sections were mounted with Fluoromount mounting medium (Cedarlane, Burlington, ON, Canada).
For 3,3’-Diaminobenzidine (DAB) amyloid-β staining, sections were treated with 0.3% hydrogen peroxide in 0.01M PBS for 10 min at room temperature. After three washes in PBS, sections were blocked as described above. Primary antibody used was anti-Aβ42. After three washes in PBS-T, sections were incubated with horseradish peroxidase-conjugated secondary antibodies (1:500, Invitrogen, Eugene, OR, USA) in PBS for 1 h at room temperature. After three additional washes in PBS-T, sections were treated with 2-solution DAB kit (Invitrogen, San Jose, CA, USA) and examined under a brightfield microscope. Reaction was stopped by rinsing under running tap water. Sections were then dehydrated with ethanol series and cleared in xylene, followed by mounting with Surgimount mounting medium.
For co-labeling using the amyloid probe K114 (see below) and immunohistochemistry, K114 staining was performed first and images were taken before immuno-labeling with 6E10 and Iba-1 because formic acid antigen retrieval disrupts the amyloid structure required for K114 binding. The same area was re-imaged and the resulting micrographs were registered and merged for colocalization analysis.
K114 amyloid staining
Staining and analysis were performed as previously described [22]. Briefly, 150μM 4,4’-[(2-Bromo-1,4-phenylene)di-(1E)-2,1-ethenediyl]bisphenol (K114, Tocris, Ellisville, MO, USA) in DMSO was diluted in a 1:1 ratio with 0.1M sodium bicarbonate buffer (pH 10.5) and applied to tissue on slides for 30 min at room temperature. The sections were then rinsed three times with sodium bicarbonate buffer and mounted with 1:1 ratio of Fluoromount and sodium bicarbonate buffer to maintain pH at 10.5. Images were acquired with a Nikon A1 confocal microscope equipped with a spectral detector. Spectral images were imported into ImageTrak (http://www.ucalgary.ca/styslab/imagetrak, written by P.K.S.) for processing and analysis.
Quantification of plaques
To quantify the number of plaques after acute injury in 27-week-old 5xFAD mice, the non-spectral K114 staining method was used to label amyloid plaques [26]. Briefly, spinal cross-sections 2 mm rostral to the lesion were incubated in 1.25 mM K114 dissolved in 40% ethanol/25 mM NaOH: PBS in 1:1 ratio for 30 min at room temperature. They were then differentiated in saturated lithium carbonate and washed in 70% ethanol. The sections were mounted with Fluoromount mounting medium and scanned on a slide scanner with a blue filter (Olympus, VS210-S5, Tokyo, Japan). At least 5 cross-sections from each animal were analyzed (n = 11 in naïve mice, n = 5 in 3- day post injury mice and n = 9 in 7- day post injury mice). Using this staining process, K114 appears mostly in the amyloid plaque core (Supplementary Figure 4). All plaques in each spinal section were counted. The same staining protocol was used to examine the presence or absence of plaques in cross-sections and longitudinal sections from different level of spinal cords, in naïve and injured 5xFAD mice.
Quantification of neprilysin fluorescence intensity
To quantify the fluorescence intensity of neprilysin in GFAP expressing cells in naïve samples and 1-,3-, and 7- days post-contusion injury samples, at least six sections from each sample were labeled with neprilysin and GFAP as described above. Sections were scanned with an Olympus slide scanner at 20x magnification. Images were imported into ImageJ (https://imagej.nih.gov/ij), GFAP (green channel) expressing area was selected as a region of interest and the mean grey value of neprilysin (red channel) staining was measured. Results were normalized against naïve samples.
Quantification of amyloid-β threads
Four longitudinal cervical sections from each unilateral contusion sample (n = 3) were stained with 6E10 and goat anti-mouse (Alexa 568) as described above, then imaged on a slide scanner. Micrographs were processed to inverted black and white images in Photoshop. Slices were divided at 250 pixel intervals across the section and any threads in the white matter intercepting the slice lines were counted. White matter area was measured in ImageJ.
Statistical analysis
Data are presented as mean±standard error of the mean (SEM), Student’s t test, one way ANOVA, or two-way repeated-measure ANOVA followed by Bonferroni’s post-hoc multi-comparison correction were used to determine the statistical significance of differences among the means. A value of p < 0.05 was considered significant.
RESULTS
5xFAD background did not affect motor recovery after injury
We initially hypothesized that 5xFAD background would result in more amyloid-β production than WT due to more AβPP accumulation in injured axons. Given the toxicity of amyloid-β, we further speculated that it would impede functional recovery of 5xFAD mice after injury. Contusion injury at the T12 spinal level is a commonly used paradigm in spinal cord injury research, allowing the monitoring of hind limb recovery over time after injury. We therefore contused 5xFAD mice and their WT littermates at T12 when they were 10 weeks old, a time point at which we previously showed no plaque deposition at this spinal level in 5xFAD mice [22]. However, no difference in functional recovery was found between WT and 5xFAD mice after 2- and 4-months post injury (Fig. 1).
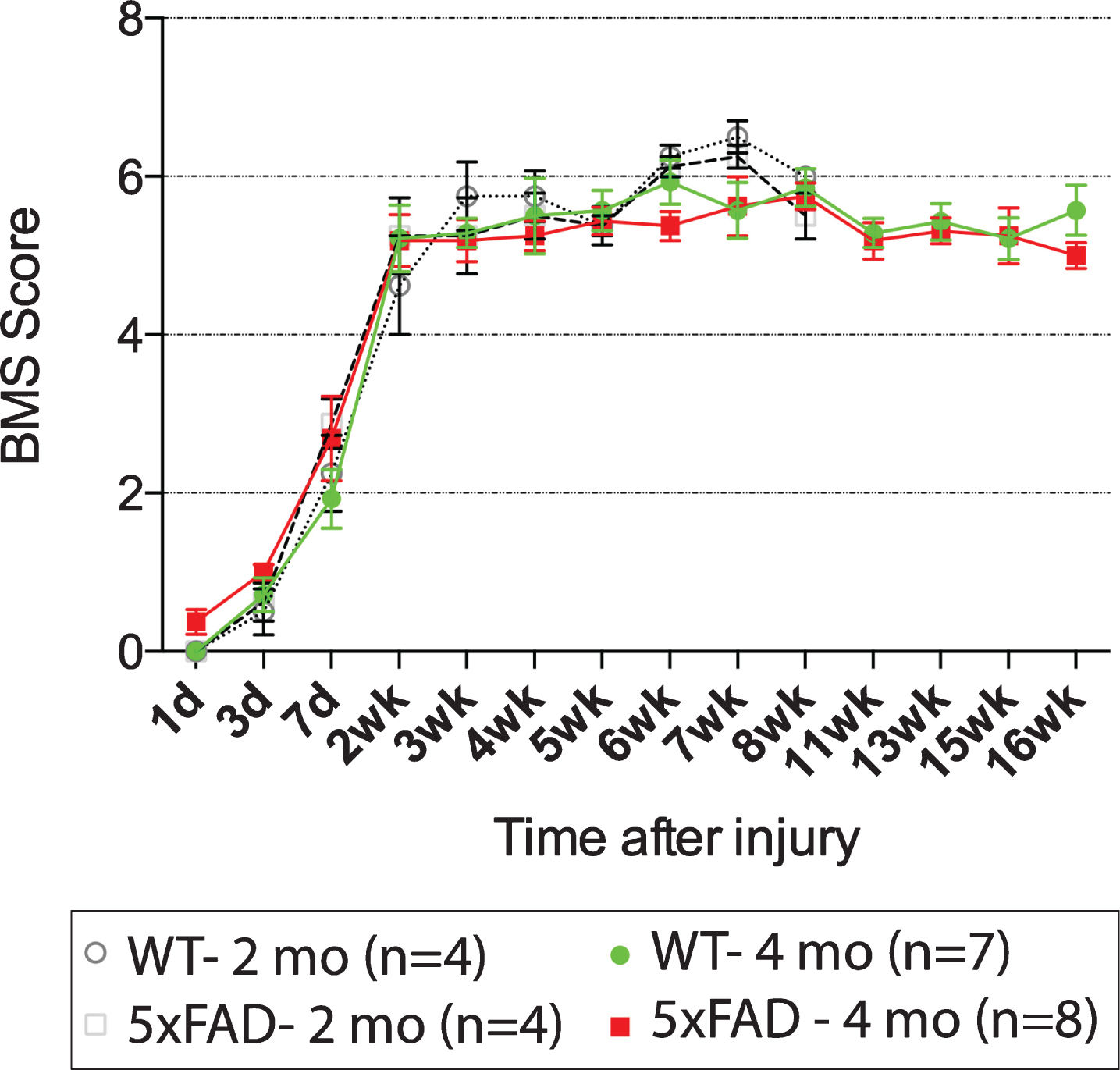
Basso Mouse Scale (BMS) for locomotion shows no difference in hindlimb functional recovery between 5xFAD mice and wild type littermates after T12 contusion injury. All mice were injured at 10-week-old and they were kept for 2 months (dotted lines) or 4 months (solid lines) for behavioral assessment. No difference between groups was found using two-way repeated measures ANOVA.
Traumatic injury reduced plaque deposition in 5xFAD mice
We removed and sectioned 5 mm of the injured low thoracic spinal cords and immuno-stained them for amyloid-β with 6E10 and Aβ42 antibodies. Compared to age-matched 27-week-old naïve 5xFAD mice which showed extensive plaque deposition, we could not find any plaques in injured WT or in the majority of injured 5xFAD mice at the T12 spinal level (Fig. 2A, B).
We then processed the remaining parts of the cords, by harvesting the 7th to 8th cervical cross sections, 3rd to 5th lumbar cross sections, and the entire thoracic cord (T1–T10) in longitudinal section from 5xFAD mice. Similar to our previously published findings [22], amyloid plaques were laden mostly in grey matter along the entire spinal cord in 27-week-old animals (Fig. 2C1 and Table 1). In contrast, only one in four mice in the 2-month post-injury 5xFAD mice showed plaques in the upper thoracic cord. More plaques were found in the upper thoracic cord in 4-month post-injury 5xFAD (Table 1) compared to the 2-month injured 5xFAD. However, a plaque-free zone with a length of 6.28±0.29 mm (n = 4) around the lesion was still observed in these 27-week-old animals (Fig. 2C2). Notably, the plaque-free zone appeared to be rostro-caudally symmetrical around the epicenter. We asked whether the absence of plaque near the epicenter was due to loss of synapses, where amyloid-β is normally produced [3]. We immunostained the sagittal sections with 6E10 and the synaptic marker anti-synaptophysin, and found dense synaptophysin staining close to lesion, in locations devoid of amyloid plaques after injury (Fig. 2D). The result is consistent with our cross-sectional staining showing normal appearing spinal cord 2 mm away from the injury epicenter (Fig. 2A3). These results indicate that the loss of plaques post-injury was not due to generalized loss of spinal gray matter at these levels, but rather, a dramatic prevention of plaque deposition present in naïve 5xFAD mice at this age.
Presence of amyloid-β plaque at different levels of spinal cord at different time points in 5xFAD mice. Data are presented as number of animals that were plaque-positive/total number of animals examined. * Level of impact injury
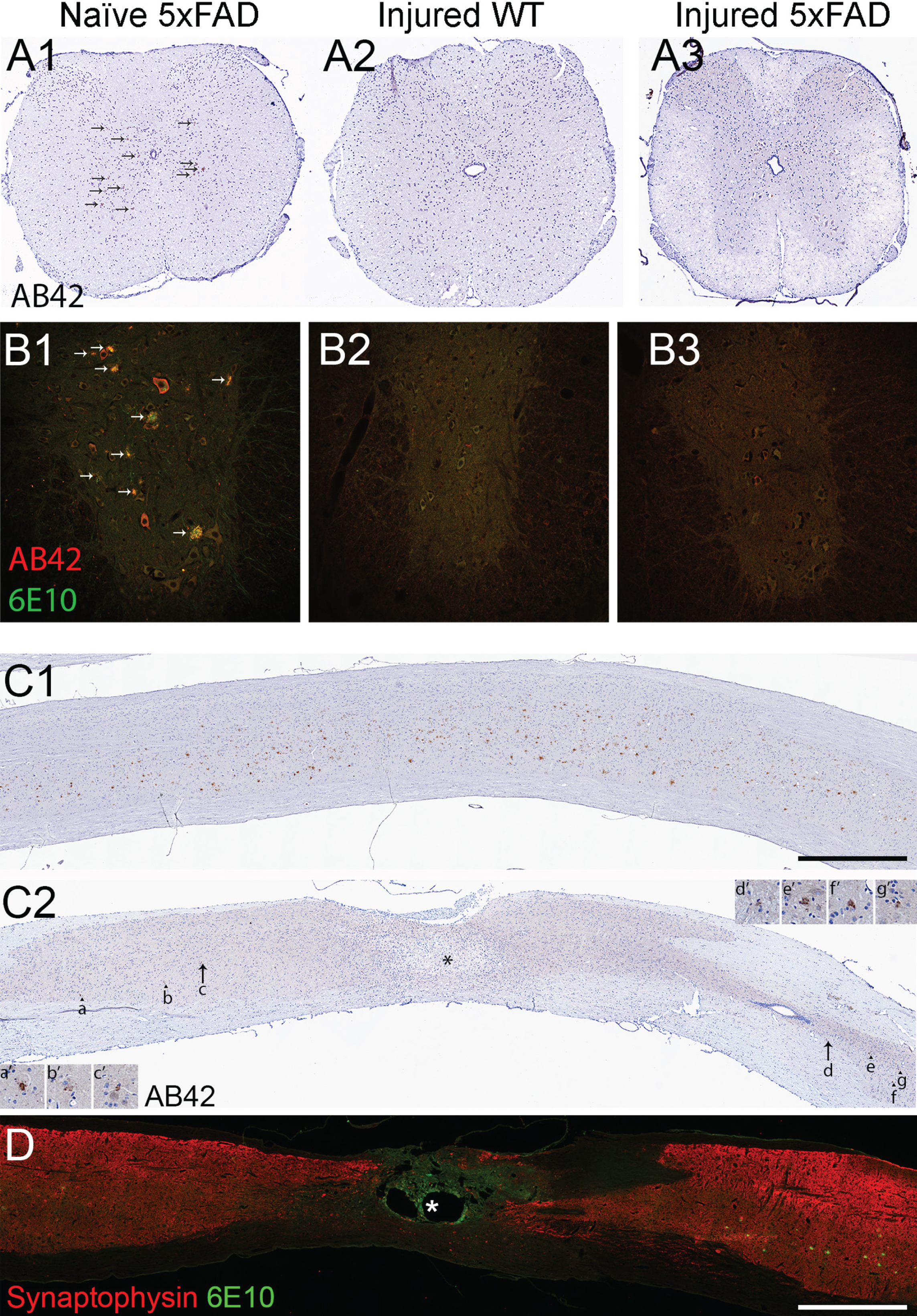
No amyloid plaque was found near the injury at 2- and 4-months post injury in the 5xFAD cord. A) Representative brightfield micrographs showing brown deposits in spinal cross sections immuno-labeled with anti-amyloid-β1–42 (Aβ42) at ∼12th thoracic level in 27-week-old naïve 5xFAD mouse (A1). In contrast, no amyloid-β was seen 2 mm rostral to the lesion, a relatively intact region without much astrocytic scar, in 4-months post-injured wild type cord as expected (A2), nor in 5xFAD cord at the same time point and location (A3). Arrows in A1 point to amyloid plaques. No plaque is found in A2 and A3. B) Representative confocal micrographs showing fluorescently labeled spinal cross sections immuno-stained Aβ42 (red) and Aβ1–16 (clone 6E10, green) at ∼12th thoracic level in 19-week-old naïve 5xFAD mouse (B1); 2 mm rostral to the lesion in 2-months post-injured wild type (B2) and 2 mm rostral to lesion in 2-months post-injured 5xFAD mouse (B3). Ventral horns are shown in B1-3. C) Representative brightfield micrographs showing DAB and hematoxylin stained spinal sagittal thoracic sections immuno-labeled with Aβ42 in 27-week-old naïve 5xFAD mouse (C1) and 4-months post-injured 5xFAD mouse (C2). Numerous amyloid plaques were observed homogeneously distributed in naïve 5XFAD grey matter while only a few plaques (arrowheads) were observed in the injured sample. Arrows in C2 mark the plaques closest to the lesion epicenter (asterisk). All plaques (a-g) are magnified in their corresponding insets (a’-g’). D) Another 4-months post injured 5xFAD mouse sample immuno-labeled with synaptophysin (red) and 6E10 (green) shows dense synaptophysin staining near the lesion epicenter (asterisk), suggesting it is not a lack of neuronal elements that was responsible for the absence of amyloid-β plaque near the lesion. Scale bar in C and D = 1 mm.
Since thoracic cord gray matter normally has lower plaque density [22], we asked whether deposition is also prevented in normally plaque-dense cervical regions. We therefore impacted the spinal cord at the seventh cervical segment with a modified unilateral contusion protocol when the 5xFAD mice were 6 weeks old, an age at which no plaques are found at any level (Table 1). We harvested the tissue 2 months after the injury. Stacked images consisted of four longitudinal cervical sections co-labeled with 6E10 and the microglial marker Iba-1, showed that the penumbral region around the impact site was essentially devoid of all plaque, compared to naïve age-matched cord that was replete with amyloid-β deposits (Fig. 3), despite robust synaptophysin signal at that region (Supplementary Figure 1).
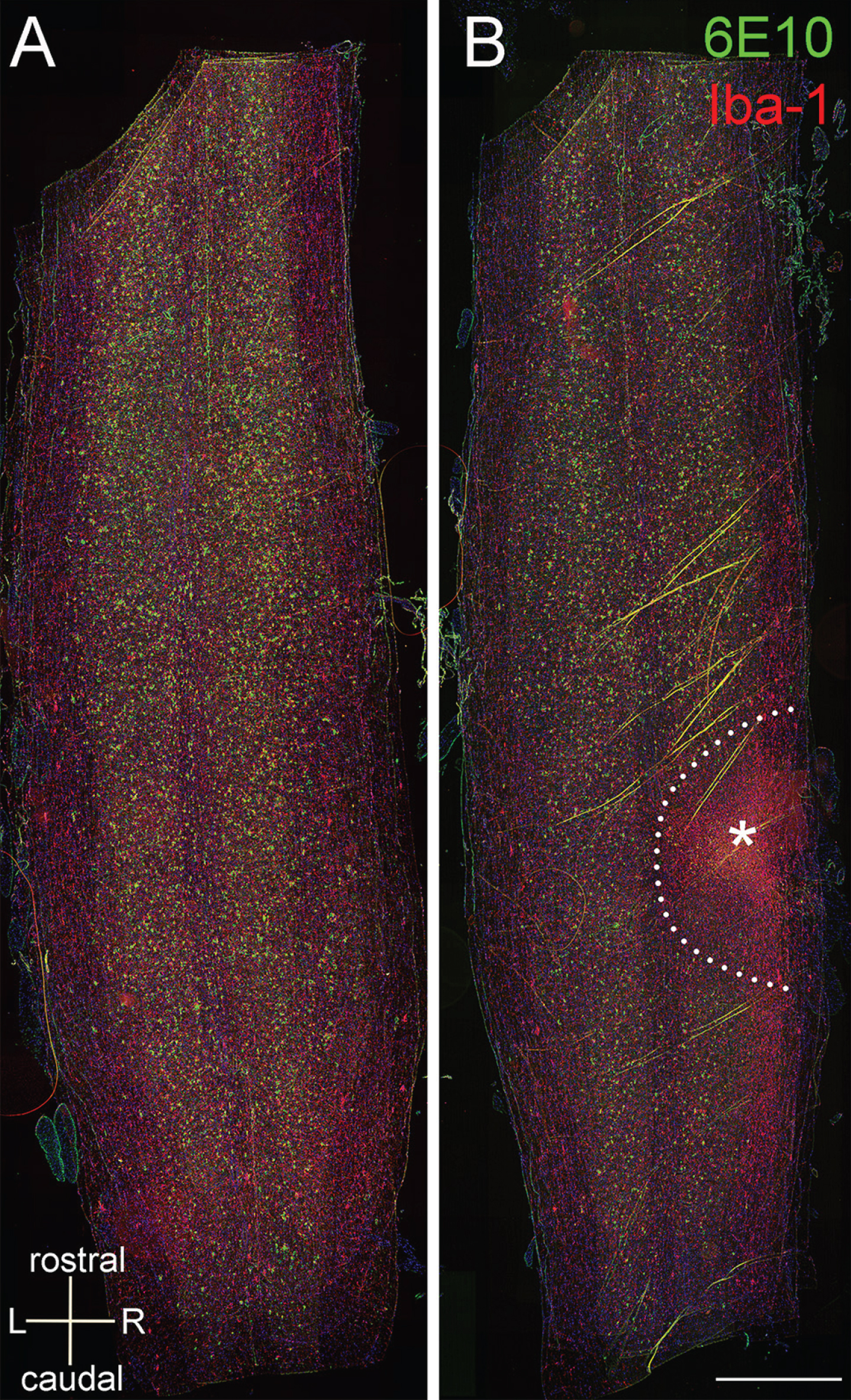
No amyloid plaque was found in the penumbral region 2 months after unilateral contusion at 7th cervical cord in 5xFAD mice. The injury was induced when the mice were 6-week-old when no plaque is found in the cervical cord. A stack of 4 cervical longitudinal sections immuno-labeled with 6E10 (green) and Iba-1 (red) show numerous amyloid deposits in 14-week-old naïve 5xFAD mouse (A) and 2-months post-injured 5xFAD mouse (B). The stainings inside the lesion (asterisk) are probably autofluorescence therefore they are positive for both red and green. The plaque free zone is outlined in dotted line in B. Scale bar = 1 mm.
Taken together, these results suggested that before appearance of plaque, injury either modified the extracellular environment or cellular activities to prevent deposition of future plaque around the lesion.
Clearance of pre-existing plaques after injury
While the above results convincingly showed that injury prevented future plaque deposition, we then asked whether trauma would also promote clearance of plaques already present at older ages. We therefore impacted 5xFAD mice at 27 weeks of age when extensive plaques are present at all cord levels [22]. Analysis of plaque number by distance away from the lesion epicenter after labeling with the amyloid probe K114 showed that there was a substantial time-dependent reduction of plaque density (Fig. 4). Quantitative analysis of plaques in cross-sections 2 mm rostral to the lesion, where the cord was not occupied by scar tissue as in the lesion core, revealed a significant reduction in the number of plaques to ≈1/3 of control density as early as 7 days after impact.

Quantitation of K114 labeled plaques in naïve, 3- and 7-day post injured 5xFAD mouse spinal cross sections. Injury was induced in 27-week-old 5xFAD mice. Statistical analysis of plaque number at 2 mm rostral to lesion epicenter in 3- and 7-day post injured mice against naïve 5xFAD mice show a significant reduction at 7 days after injury. The outlier with >20 plaque number in day 7 group was omitted in the analysis after Grubbs’ test. ***p = 0.0002. R2 and R1: 2 mm and 1 mm rostral to lesion core; C1:1 mm caudal to lesion core.
Expression of neprilysin in astrocytes and microglia/macrophages
The clearance promoted by traumatic injury of pre-existing amyloid-β deposits from the cord was striking and unexpected. We then explored possible mechanisms by which these abnormal deposits could be removed by examining the expression of neprilysin, a metallo-endopeptidase responsible for amyloid-β degradation [27–29]. In the spinal cord, this enzyme was found to be expressed in motor neurons, the intermediolateral cell column in the thoracic cord, Schwann cells and occasionally in astrocytes in naïve animals (Supplementary Figure 2). We asked if the upregulation of neprilysin could account for amyloid clearance. No neprilysin staining was observed in astrocytes or microglia in naïve mouse cord samples or 1 day after the injury in 27-week-old 5xFAD mice (Figs. 5 and 6C). In contrast, at 3 and 7 days after injury, increased neprilysin immunoreactivity was observed in GFAP-positive astrocytes (Fig. 6A, C) close to the contusion epicenter. Co-expression of neprilysin and GFAP returned to basal level in region further away, i.e., almost absence of neprilysin in astrocytes approximately 2 mm rostral and caudal to lesion epicenter. Most of the Iba-1 positive microglia were neprilysin negative at these time points, with only a few weakly stained (Fig. 6B and Supplementary Figure 3). Interestingly, when we examined the 4-month post-injured samples (impact was performed at 10 weeks of age), strong neprilysin expression persisted only at the lesion core; however, we found most of the neprilysin staining was in Iba-1 positive cells instead of GFAP positive cells, suggesting a downregulation of neprilysin in astrocytes and modest upregulation in microglia/macrophages in the chronic phase of injury (Supplementary Figure 4).
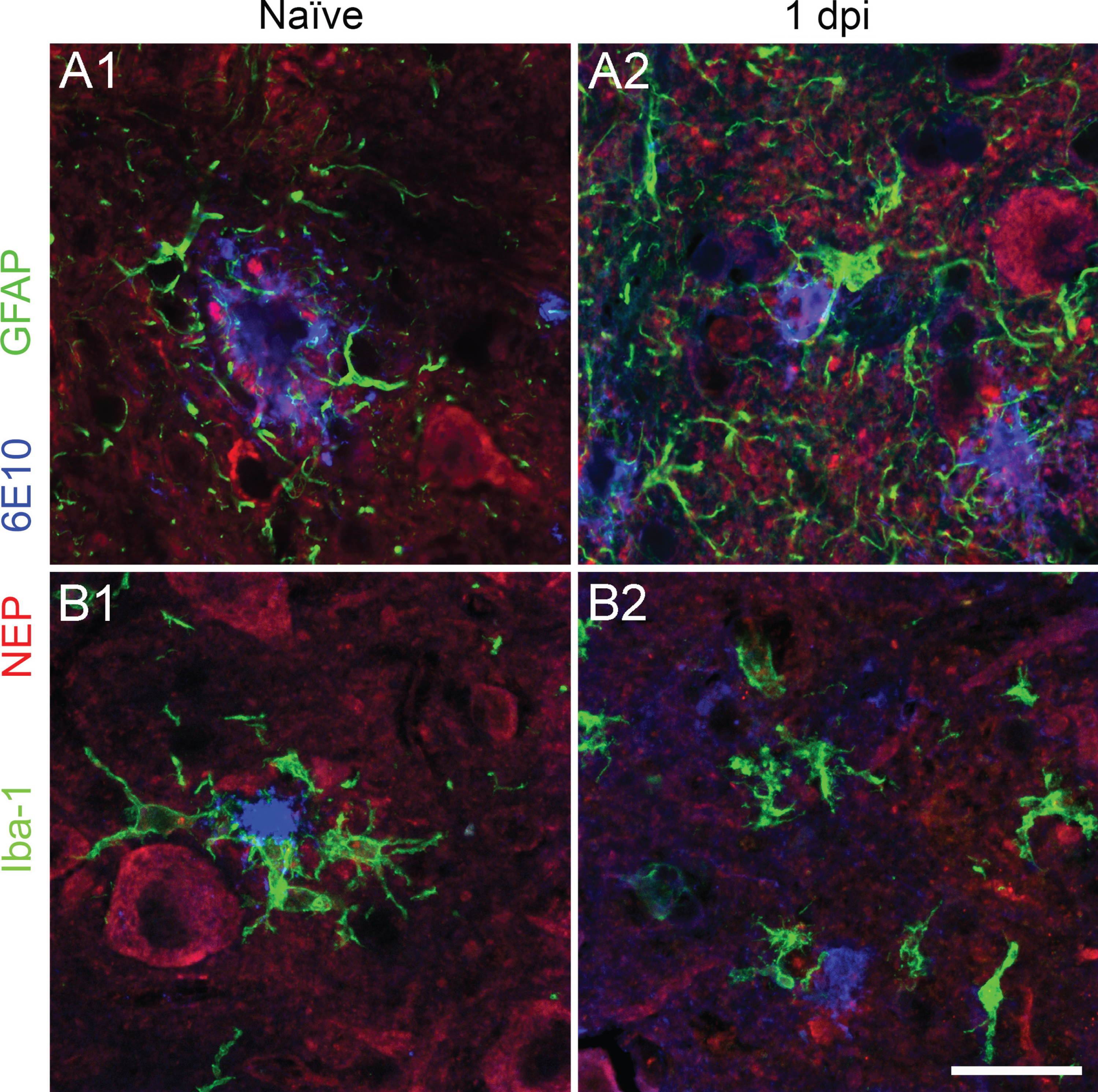
Neprilysin is not expressed in astrocytes or microglia in naïve or 1-day post-injured 5xFAD cord. Representative fluorescent labeled spinal cord cross sections show that neither astrocyte (A, green) nor microglia (B, green) express neprilysin (red) in naïve and 1 day after injury in 5xFAD mice. Motor neurons and dystrophic neurites around the 6E10 positive amyloid plaque (blue) are neprilysin positive. Scale bar = 25μm.
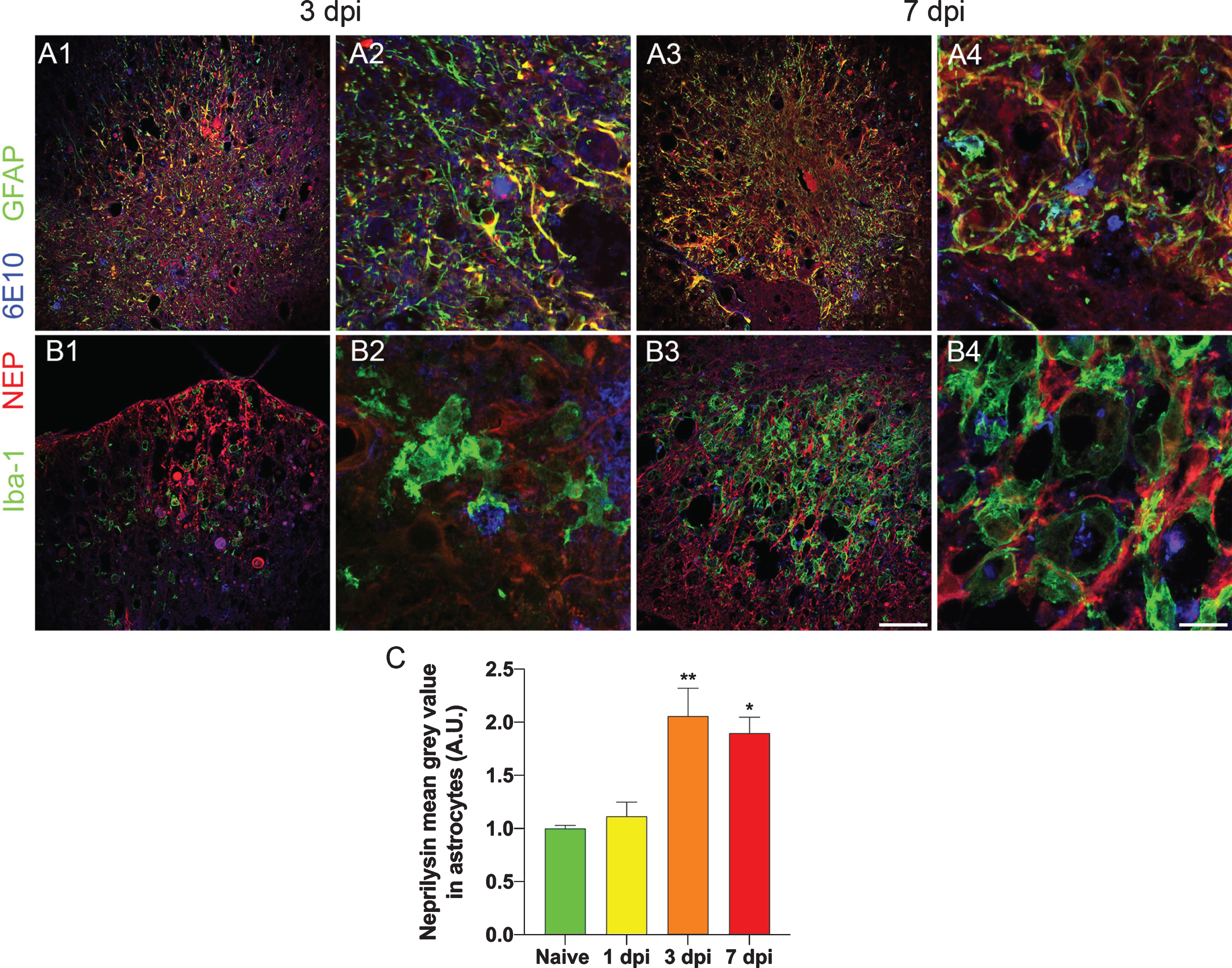
Neprilysin is upregulated in astrocyte starting at 3 days after injury in 5xFAD. Low magnification of 6E10 (blue), neprilysin (red), and GFAP/Iba-1 (green) stained spinal sections from 3-day (A1, B1) and 7-day (A3, B3) post injured 27-week-old 5xFAD mice. Higher magnification micrographs are shown in A2, A4, B2, and B4. Results show that astrocytes, but not microglia, upregulate neprilysin at 3 and 7 days after injury. Quantification of neprilysin staining intensity in GFAP expressing cells revealed significant up-regulation of neprilysin intensity at 3 and 7 days after injury (*p < 0.05; **p < 0.01 versus naïve). A.U., arbitrary unit; dpi, days post injury. Scale bar for low magnification micrographs = 100μm. Scale bar for high magnification micrographs = 20μm.
Role of microglia in clearance
Microglia have been implicated in removing amyloid-β plaque by phagocytosis [30, 31]. Indeed, we found morphological changes in microglia starting at 1 day after injury, showing shorter and stouter processes (Fig. 5B2), a typical response to injury. At 3 days after injury, many assumed an amoeboid shape and fewer processes were contacting plaques (Figs. 6B2 and 7B2). Some Iba-1 positive cells were seen engulfing the plaque (Fig. 7B3 arrowhead). At 7 days after injury, the spinal cord cross section at the lesion was mostly occupied by Iba-1 positive cells. We found many 6E10 inclusions in these cells at the epicenter. To verify that they are amyloid species, we stained the sections with K114. In naïve samples from 27-week-old 5xFAD mice, K114 faithfully labeled amyloid plaques positive for 6E10 (Fig. 7A1-2). However, the staining pattern varied slightly in that K114 brightly labeled the core whereas 6E10 also labeled the rim of the plaque in the naïve condition (Supplementary Figure 5). At 3 days after injury, some plaque-specific K114-positive inclusions were seen inside Iba-1 positive cells (Fig. 7B1–3 arrowheads) and at 7 days after injury, more K114-positive inclusion, also positive for 6E10, was found inside Iba-1 positive cells. The presence of 6E10 and K114 positive staining found within microglia/macrophage suggests that bigger amyloid plaques might be broken down and engulfed by these cells for clearance.
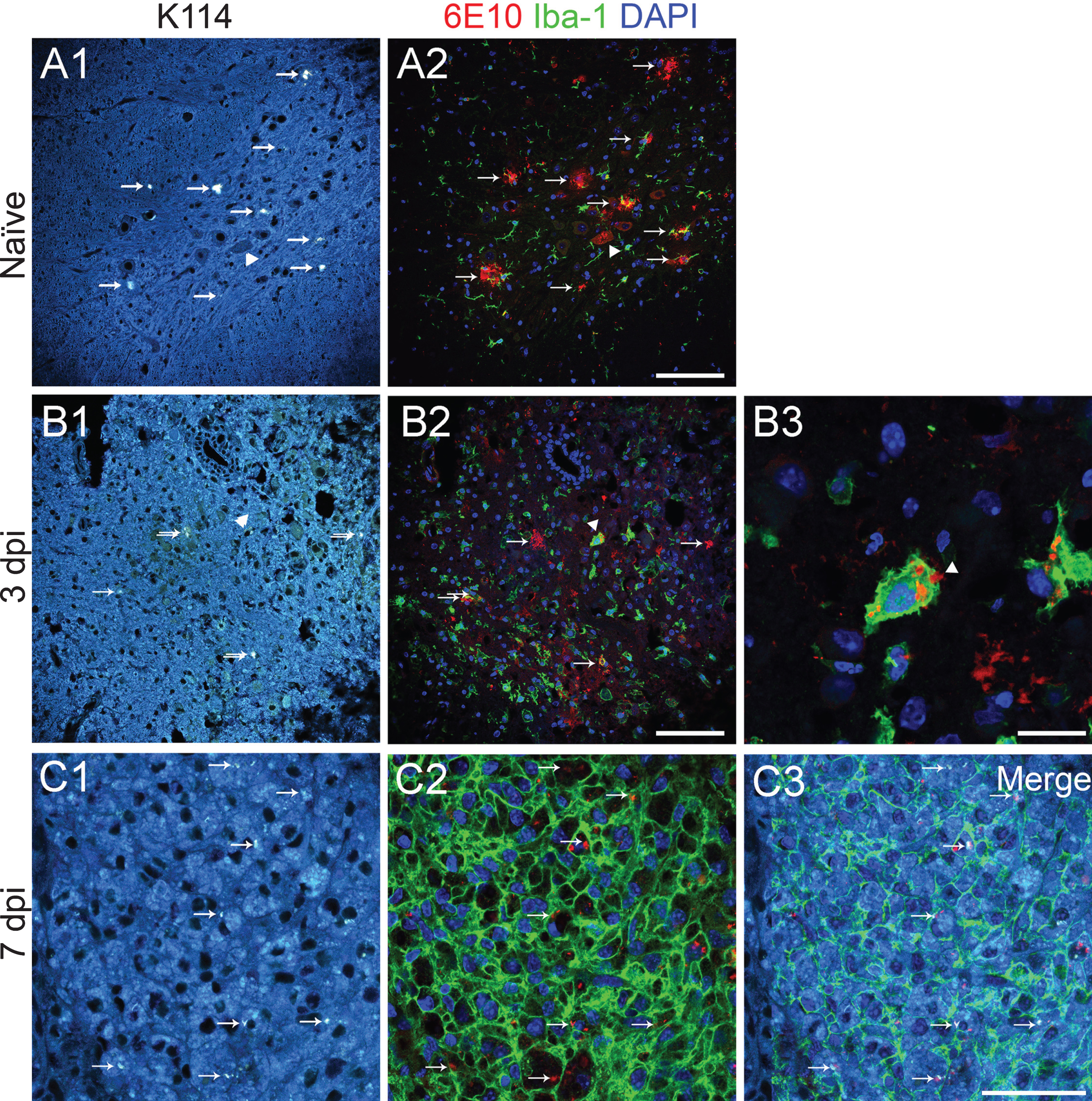
6E10 positive inclusions inside microglia are K114 positive. True color images (A1, B1, C1) show amyloid-β plaques (arrows). After K114 spectral images were taken, sections were then treated with formic acid and stained with 6E10 (red) and Iba-1 (green) and counterstained with DAPI (blue). Results show that K114 labeling matches 6E10 immunostaining in 27-week-old naïve 5xFAD mouse sample. B) At 3 days after injury, some plaques are observed inside Iba-1 positive cells (arrowed cell is enlarged in B3). C) At 7 days after injury, many 6E10 and K114 positive inclusions are found inside Iba-1 positive microglia/ macrophage. It suggests that the engulfed amyloid plaques still maintain their original confirmation inside these immune cells. Scale bar for A1–A2, B1–B2 = 100μm; scale bar for B3 = 20μm; scale bar for C1–C3 = 50μm.
Reduction of thread-like amyloid deposits
We recently identified thread-like deposition of amyloid-β positive material in the peri-axonal space of spinal white matter axons, which likely represent the earliest pathology that is a precursor to larger plaque accumulation in white matter [22]. To examine the effects of injury on potential clearance of these very early pathological amyloid deposits- which can be readily identified in white matter tract [22], we hemisected the dorsal right side of the 12th thoracic spinal cord in 27 week old 5xFAD mice (n = 3) and harvested them at 7 day post injury. Hemisection resulted in a localized damage only on the injured side, allowing the use of the contralateral side for internal comparison. We then sectioned them longitudinally, labeled them with 6E10, and compared the number of threads on both sides (Fig. 8A). All samples (n = 3) showed a reduction of these deposits by a factor of 2 at the ipsilateral side compared to the contralateral side (14.8±3.8 versus 30.3±4.1, p = 0.025, Wilcoxon rank test, Fig. 8B).
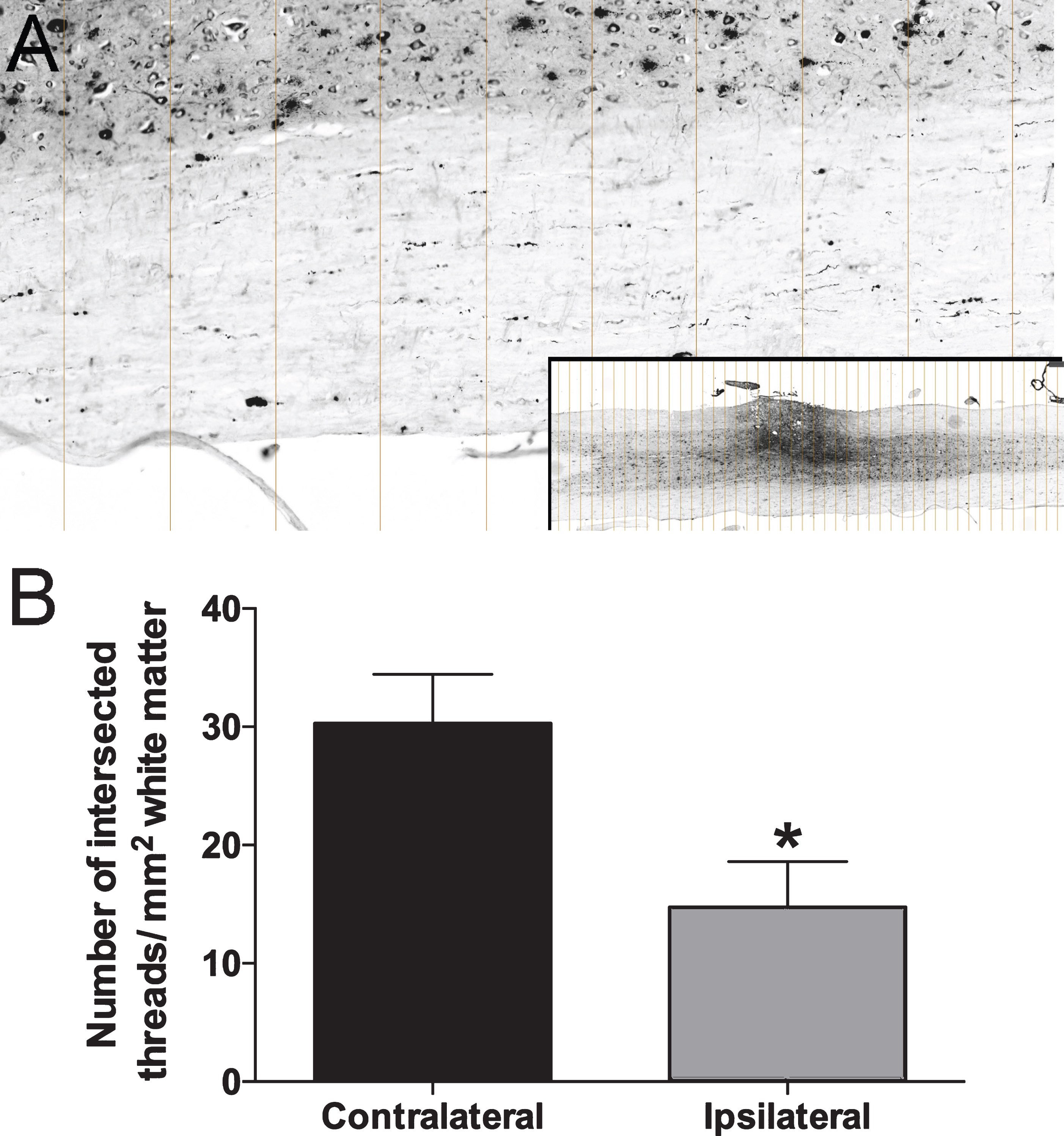
Reduction of amyloid-β thread after injury. To minimize variations between animals, we analyzed the amount of thread in dorsal hemisectioned 5xFAD mice by comparing ipsilateral lesion side with contralateral non-lesion side. A) Entire longitudinal sections were imaged with slide scanner (inset in A) and micrographs were sliced at 250 pixel interval in Photoshop. All threads in white matter intercepting with the lines were counted. The area of white matter was measured with ImageJ. B) Quantitative results for 3 samples show significant reduction of number of amyloid-β threads.
DISCUSSION
A summary of our experiments is shown in Table 2. The main finding of our study is a dramatic reduction in amyloid plaque burden in the 5xFAD mouse spinal cord after focal traumatic injury. This reduction was manifest as either a prevention of later amyloid deposition if the injury occurred before the age at which plaques appear spontaneously, or a rapid clearance of pre-existing amyloid plaques and threads if mice were injured at a later age when plaque pathology was already well established. Quite remarkably, the latter occurred within days of the injury (Fig. 4B). Traumatic spinal cord injury triggers a cascade of inflammatory events that last for weeks to months [32]. One of the major cell types involved are microglia which seem to exhibit dichotomous roles after injury [33]. It has been shown that they release pro-inflammatory cytokines and reactive oxygen/nitrogen species that cause cell death, and on the other hand, they phagocytose debris and differentiate into anti-inflammatory phenotype which is beneficial for tissue repair [34–38]. In 5xFAD or other AD mouse models, microglia are already activated due to amyloid plaque deposition [23, 40]. In vivo imaging of microglial interaction with amyloid-β plaques in Iba-1-GFP and APP/PS1 hybrid mouse showed that once microglia make contact with a plaque, they tend to engage with the same plaque and in some cases, internalize part of the plaque, thus reducing its size [41]. This interaction also limits the spread of neurotoxic amyloid species [42]. Despite the ability of microglia to encapsulate and internalize plaque amyloid, plaques are persistently formed and they slowly grow in size [43], suggesting that the plaque clearance process is inefficient. The pro-inflammatory extracellular milieu may account for their limitation in their ability to clear plaque. For example, incubation of an activated microglial cell line BV-2 with pro-inflammatory cytokines such as interleukin-1β inhibits phagocytosis of fibrillar amyloid-β 1–42 [44]. In contrast, drastic change in cytokine composition and antibody production [45] after traumatic injury may reactivate microglia to remove plaque. It has been shown that administration of exogenous monoclonal antibodies against amyloid-β reduces the number of small or diffuse plaques in 13-month-old PDAPP mice through microglial phagocytosis [46]. Our study is in line with other studies that specifically looked at extracellular plaque deposition and found a regression of amyloid deposits after traumatic brain injuries [18, 19], suggesting the injury response is similar in the spinal cord and brain.
Different types of spinal cord injuries and parameters used in the study
*no amyloid plaque was found at cervical cord at this age. **extensive amyloid plaques were found at thoracic cord at this age
Another major cell type that might help clear plaques is the monocyte derived macrophage. Although they are indistinguishable from microglia in terms of morphology and immunohistochemical markers, new data using RNA sequencing showed that they not only express a common pool of genes but also a distinct set of genes by individual cell types [47]. Irradiated chimera reconstituted with GFP-tagged bone marrow cells or knock-in transgenic mice expressing EGFP in lysosome (lys-EGFP-ki mouse) provide a means to distinguish resident microglia from infiltrated hematopoietic stem cell derived macrophages [48–50]. These studies showed that blood borne macrophages entered the lesion site at 3 day after spinal cord injury [48, 50], possibly through the choroid plexus in the brain [49]; similar timing of macrophage infiltration is seen after traumatic brain injury. Based on these studies, we speculate that the early modest reduction of amyloid-β at 3 days after injury is probably mediated through activated microglia while the robust clearance at 7 days is mediated through infiltrating macrophages. It is intriguing to find 6E10 positive inclusions inside microglia/ macrophage that are still K114 positive indicative of persistent beta sheet-rich amyloid assemblies [26, 51]. This suggests that Iba-1 positive cells (microglia/macrophages) are able to remove components of amyloid-β from larger plaques, but may have difficulty digesting the relatively protease-resistant beta sheet-rich aggregates.
Our results suggested that the concerted efforts of astrocytes and microglia/macrophages may account for efficient clearance. We identified an upregulation of neprilysin in astrocytes which may help degrade and remove amyloid plaque. In addition to insulin-degrading enzyme, presequence peptidase, endothelin converting enzyme, angiotensin converting enzymes and others, neprilysin is one of the major amyloid-β degrading enzymes [52, 53]. Targeted disruption of neprilysin in mice resulted in enhanced amyloid-β 1–40 and 1–42 in various brain regions including cortex and hippocampus [27]. In contrast, overexpression of neprilysin or enhancing its activity in AD mouse models reduced plaque burden and mitigated memory impairment [54–57]. Similar to our finding in naïve cord samples, no neprilysin staining was found in GFAP labeled astrocytes (Fig. 5A1). The upregulation of neprilysin in plaque-engaging astrocytes starting at 3 days post injury matches the onset of plaque removal, suggesting that these cells might play a key role in plaque clearance. The increase of neprilysin expression seems to be mediated by apolipoprotein E (ApoE) [58]. It has been reported that ApoE protein level is upregulated in astrocytes and microglia as early as 4 days after spinal cord injury in C57BL/6 mice. And it has been shown that primary cultured astrocytes incubated in amyloid-β will only upregulate neprilysin in the presence of ApoE [59]. These results are consistent with our findings that plaque-engaged astrocytes do not normally express neprilysin but expression is observed only after injury. Similar upregulation of ApoE protein and amyloid clearance was also found after traumatic brain injury in APP/PS1 mice [60]. Our immuno-staining also revealed a weak expression of neprilysin in some Iba-1 positive microglia/macrophages early after injury, suggesting these cells are also able to degrade and remove plaque autonomously. Interestingly, neprilysin expression is more prominent in Iba-1 positive cells in the lesion core at 4 months after injury; the location of these cells at the epicenter suggested that they are likely to be blood borne macrophage [48], therefore it is possible that the Iba-1 positive/neprilysin positive cells detected early after injury are monocyte derived macrophages.
Our most intriguing observation was 5xFAD spinal cord at a large distance (>half a centimeter) from the lesion core that remained free of plaque even at 27 weeks of age, a time point when significant plaque load is normally found in uninjured 5xFAD mice. In this case, the animals were injured at 10 weeks of age before any plaques were deposited in the thoracic cord. The absence of plaques at or around lesion site suggested that the microenvironment was rendered unfavorable for plaque formation. Further studies are required to identity the factor(s) that prevented plaque deposition. It is reasonable to assume that the injured supraspinal axons retracted from the impact site so fewer plaques were formed near the lesion. Another possible explanation is that the reduction of amyloid-β threads after injury may have resulted from the reduction of electrical activity as previously suggested [22]. We showed that these threads form in the axon and accumulate in the peri-axonal space, and can be potential pre-plaque structures. It is possible that these structures were exposed due to demyelination after injury [61], recognized and removed by microglia/ macrophages, thus reducing the formation of eventual plaques. Indeed, we found a reduction in the amount of threads after hemisection in the white matter (Fig. 8). It is noteworthy that hemisection is a simple axonal injury model compared to the more traumatic and clinically relevant contusion model that we used in earlier parts of our study; its limitation notwithstanding, this type of injury is highly reproducible and provides a contralateral internal control for quantification of threads. The signals responsible for plaque reduction at remarkably long distances from the injury focus remain a mystery, and deserve further investigation (Table 1). However, given the robust inflammatory response known to occur in the spinal cord after trauma [62, 63], together with the robust upregulation of neprilysin expression in innate immune cells (astrocytes and microglia/macrophages) we show here, hints at innate immune signaling as a key factor in promoting the exceptional clearance of plaques we report in the 5xFAD mouse model, which might be somewhat unphysiological given its very strong promotion of amyloid-β deposition.
Our results show that astrocytes and microglia/macrophages likely work in synergy to clear amyloid-β plaque after trauma. We propose that trauma activates astrocytic NEP which enhances break down of amyloid plaque, followed by ingestion of (partially) digested amyloid debris by microglia/macrophages. Our results are consistent with a two-pronged approach leading to plaque clearance, i.e., increasing neprilysin activity in astrocytes to help break down amyloid followed by stimulation of innate immune cells to ingest and dispose of the proteolyzed debris. What remains unanswered is what stimuli arise after a focal injury, operating at considerable distances and for prolonged periods of time, that promote clearance of pre-existing amyloid deposits and prevent deposition of new plaque as the mouse ages. Elucidation of mechanisms may inform new therapeutic strategies for AD and possibly other protein misfolding disorders.
Footnotes
ACKNOWLEDGMENTS
This study was supported by grants from the Dr. Frank Leblanc Chair for Spinal Cord Research, Canadian Institutes of Health Research (CIHR), Canada Research Chairs (CRC) and an Alberta Prion Research Institute (APRI) Explorations grant. All experiments on animals received ethical approval from the University of Calgary.