
Abstract
Background:
Cardiac ischemia/reperfusion (I/R) injury induces brain damage through increased blood-brain barrier (BBB) breakdown, microglial hyperactivity, pro-inflammatory cytokines, amyloid-β deposition, loss of dendritic spines, brain mitochondrial dysfunction, and imbalanced mitochondrial dynamics. Previous studies demonstrated that mitochondrial fusion promoter reduced cardiac damage from cardiac I/R injury; however, following cardiac I/R injury, the roles of mitochondrial dynamics on the brain have not been investigated.
Objective:
To investigate the effects of pharmacological modulation using mitochondrial fusion promoter (M1) in the brain of rats following cardiac I/R injury.
Methods:
Twenty-four male Wistar rats were separated into two groups; 1) sham-operation (n = 8) and 2) cardiac I/R injury (n = 16). Rats in the cardiac I/R injury group were randomly received either normal saline solution as a vehicle or a mitochondrial fusion promoter (M1, 2 mg/kg) intravenously. Both treatments were given to the rats 15 minutes before cardiac I/R injury. At the end of the reperfusion protocol, the brain was rapidly removed to investigate brain mitochondrial function, mitochondrial dynamics proteins, microglial activity, and Alzheimer’s disease (AD) related proteins.
Results:
Cardiac I/R injury induced brain mitochondrial dynamics imbalance as indicated by reduced mitochondrial fusion proteins expression without alteration in mitochondrial fission, brain mitochondrial dysfunction, BBB breakdown, increased macrophage infiltration, apoptosis, and AD-related proteins. Pretreatment with M1 effectively increased the expression of mitofusin 2, a mitochondrial outer membrane fusion protein, reduced brain mitochondrial dysfunction, BBB breakdown, macrophage infiltration, apoptosis, and AD-related proteins in rats following cardiac I/R injury.
Conclusion:
This mitochondrial fusion promoter significantly protected rats with cardiac I/R injury against brain damage.
INTRODUCTION
Acute myocardial infarction (AMI) is the leading cause of death worldwide [1]. Several forms of therapeutic management have been used in patients with AMI to reduce cardiac damage. These include administration of fibrinolysis, percutaneous intervention, and coronary artery bypass surgery [2]. All of these are aspects of reperfusion therapy. Following the recovery from AMI, patients can develop several neurological disorders including depression, anxiety, mood disorders, psychosis, and some symptoms of neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases [3, 4]. In animal models, brain damage has been observed after cardiac ischemia/reperfusion (I/R), including cognitive impairment [5] and the reduction of dendritic spine density [6, 7]. The elemental mechanisms of brain damage after cardiac I/R injury have been extensively studied. Previous studies showed that disruption of the blood-brain barrier (BBB) is one possible cause of brain damage following cardiac I/R injury [7, 8]. Cardiac I/R injury has been shown to increase brain oxidative stress [5, 7], which could decrease effective function of tight junction proteins, such as claudin 5 and Zonula occludin-1 [9], resulting in BBB breakdown. Pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1β (IL-1β), were recruited and accumulated in the brain after BBB breakdown following cardiac I/R injury [10–12]. A nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) was also activated [6], leading to microglial hyperactivity and brain inflammation [5, 13]. In physiological condition, mononuclear phagocytes in the central nervous system, including microglia, express leukocyte common antigens [14]. Leukocyte common antigens, CD11b and CD45, were found to be expressed in the microglia of the central nervous system, therefore, the co-expression of CD11b and CD45 antigens has been used as a microglial marker [14]. In addition, the upregulation of CD45 has been found in the microglia of mice with Alzheimer’s disease (AD) in response to inflammation [15]. A previous study reported that CD11b+/CD45+high cells represented the activated microglia or the infiltrating macrophages, whereas CD11b+/CD45+low cells were synonymous with resident microglia [15]. Furthermore, brain inflammation is related to the pathogenesis of AD as shown by hyperphosphorylated tau and amyloid-β (Aβ) aggregation [14].
In addition to brain inflammation, brain mitochondrial dysfunction has been proposed as another possible mechanism underlying brain damage after cardiac I/R [8]. Recently, mitochondrial dynamic imbalance was shown to be involved in several pathogeneses of the brain such as AD and stroke [15, 16]. Mitochondrial dynamics consists of two key processes, mitochondrial fission and mitochondrial fusion [8, 17–19]. The balance of mitochondrial fission and fusion is necessary for the maintenance of mitochondrial function, apoptosis, mitochondrial biogenesis and distribution, and complementation of mitochondrial DNA and cell division [17]. Our previous study reported that cardiac I/R injury causes a reduction in mitochondrial fusion protein, Mfn2, whereas mitochondrial fission protein, Drp1, was not altered in the brain [8]. Therefore, mitochondrial dynamic modulators could be a potential therapeutic strategy to reduce brain damage in cardiac I/R injury. Both mitochondrial fission inhibitor (Mdivi-1) and mitochondrial fusion promoter (M1), the pharmacological mitochondrial dynamic modulators, showed beneficial effects for the heart by attenuating cardiac damage and improving cardiac function during cardiac I/R injury [20–22]. However, the effects of mitochondrial dynamics modulators, particularly M1, which are the cell-permeable phenylhydrazone compounds that dose-dependently induced mitochondrial elongation [23], on the brain pathology following cardiac I/R injury have never been explored. Therefore, we hypothesized that the modulation of mitochondrial dynamics by a mitochondrial fusion promoter attenuates the brain injury via restoration of the balance of brain mitochondrial dynamics, reducing brain mitochondrial dysfunction, brain inflammation, and AD related protein expression in rats following cardiac I/R injury.
MATERIAL AND METHODS
All experimental protocol in this study was permitted by the Institutional Ethics Committee for animal research, Faculty of Medicine, Chiang Mai University, Thailand (permit no. 05/2560), under the ARRIVE guidelines for reporting data from research using animals. Twenty-four male Wistar rats (weight 250–300 g.) were used, and they were obtained from the Nomura Siam Company, Bangkok, Thailand. Rats underwent either a sham operation (n = 8) or cardiac I/R (n = 16). In the cardiac I/R group, rats were brought to 30 min of cardiac ischemia and 120 min of reperfusion as previously described [20]. Rats in the cardiac I/R group were given their assigned intervention (vehicle; normal saline solution or M1, 2 mg/kg, intravenously; [20]) at 15 min prior to cardiac I/R. The single concentration of the M1 used in the present study was selected following the outcome of a previous study. That study showed that 2 mg/kg of M1 effectively reduced myocardial infarct size and prevented cardiac mitochondrial dysfunction following cardiac I/R [20]. In the sham operated rats, the heart was exposed by thoracotomy, and the left anterior descending coronary artery (LAD) was recognized, but it was not ligated. The cardiac ischemia was done by ligating the LAD at 2–3 mm distal to its division. A successful ischemia was confirmed by ST segment elevation on an electrocardiogram [20]. Reperfusion was done by loosening the knot, and rats were subjected to reperfusion for 120 min, then the rats were killed by decapitation. The experimental protocol is shown in Fig. 1. The diagram showing the areas of the brain used for specific experiments is shown in Fig. 1B. The left hemisphere was used for microglial isolation to determine the number of CD11b+ and CD45+ cells. The right hemisphere was used for mitochondrial isolation and western blotting to determine brain mitochondrial function, mitochondrial dynamics protein expression, apoptosis, and AD related proteins including Aβ, amyloid-β protein precursor (AβPP), phosphorylated tau protein (p-Tau), and tau protein (tau).
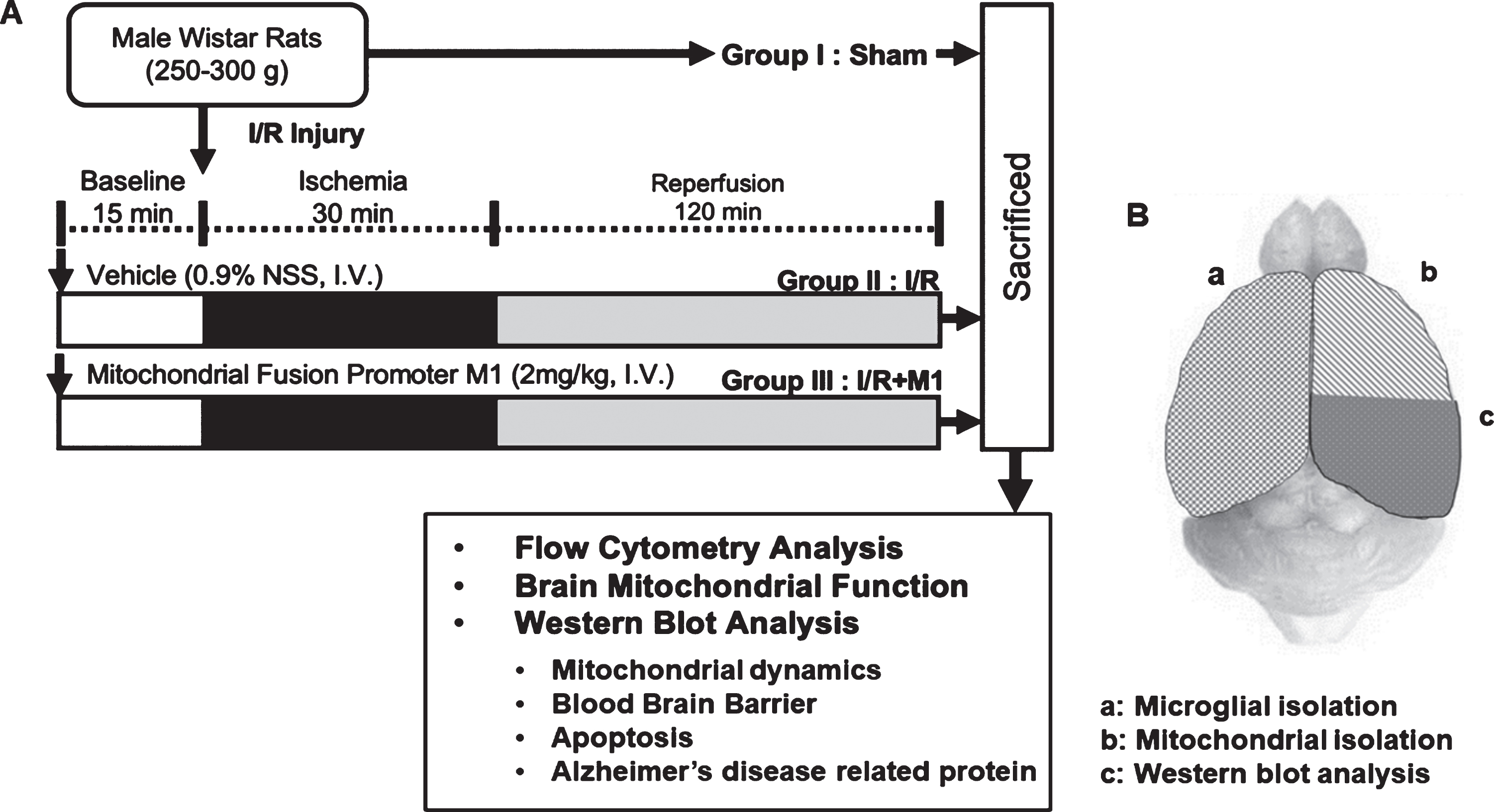
A) Experimental diagram, and B) the areas of the brain used for biochemical analysis: (a) left hemisphere was used for microglial isolation, (b) right hemisphere was used for mitochondrial isolation, and (c) western blotting.
Cardiac I/R protocol
The rats were anesthetized with a combination of Zoletil (50 mg/kg, intramuscular injection) and Xylazine (0.15 mg/kg, intramuscular injection). The anesthetic depth was confirmed by absence of eyelid reflex, pedal withdrawal reflex, and tail pinch reflex. Then, the tracheostomy was done under controlled ventilation with a rodent ventilator (CWE-inc, PA, USA). The left thoracotomy was done at the 4th–5th intercostal space, and the heart was exposed. The LAD was recognized, and a 2–3 mm ligation was done distal to its division for 30 min, then the knot was loosened to allow myocardial reperfusion for 120 min.
Microglial activity determination
The brain was removed and homogenized at 200 rpm in Hanks’ Balance Salt Solution. The homogenate was filtered through a 0.2μm filter, then the filtrate was centrifuged at 500 g, at 25°C for 6 min. The pellet was mixed with 70% percoll in PBS, and it was overlayed by 50% percoll in PBS, 30% percoll in PBS, and PBS, respectively. The layers were centrifuged at 2000 g, at 25°C for 20 min, and the microglia were collected at the interface between the 70% and 50% Percoll layers [6]. Microglia were stained with anti-FITC-CD11b (1:20 dilution, BD pharmigen) and anti-PE-CD45 (1:20 dilution, BD pharmigen) then the identification of CD11b and CD45 was done by flow cytometry (FACS Celesta, BD bioscience, USA). A co-stain of CD11b+ and CD45+ cells was considered as microglia. The percentage of CD11b+/CD45+ high presented cells was considered as equivalent to active microglia (infiltrated macrophage), and the percentage of CD11b+/CD45+ low presented cells was considered as resident microglia [6, 25].
Brain mitochondrial function determination
The brain mitochondria were isolated as described in previous studies [8, 11]. The brain was homogenized in MSE solution (MSE is a buffer containing Mannitol (225 mM), Sucrose (75 mM), EGTA (1 mM), HEPES (5 mM), and BSA (1 mg/ml)) supplemented with Nagarse (0.5 mg/ml). The homogenates were centrifuged at 2000 g for 4 min, and 12000 g for 9 min. The brain mitochondrial pellet was resuspended in MSE solution containing Digitonin (0.2 mg/ml), and the mitochondrial pellet was washed at 12000 g for 11 min. Then, the bicinchoninic acid assay kit (Sigma-Aldrich, MO, USA) was used to determine the levels of mitochondrial proteins. 0.4 mg/ml of isolated brain mitochondria were used to measure brain mitochondrial function.
Brain mitochondrial Reactive Oxygen Species (ROS) determination
ROS production was identified by using DCFH-DA (2′,7′-Dichlorodihydrofluorescein diacetate) dye. Brain mitochondria were incubated with 2μM DCFH-DA at 25°C for 20 min. The DCF intensity was measured at λex 485 and λem 530 nm via a fluorescent microplate reader. An increased DCF intensity represents a high level of brain mitochondrial ROS [8].
Brain mitochondrial membrane potential determination
Brain mitochondrial membrane potential alterations were measured using 5,5′,6,6′-tetrachloro-1,1′,3,3′- tetraethylbenzimidazolcarbocyanine iodide (JC-1) dye. Isolated brain mitochondria were incubated with 310 nM of JC-1 dye for 15 min at 37°C. Red (485/590 nm, JC1 aggregated form) and green (485/530 nm, JC1 monomer form) fluorescence intensity of JC-1 were determined using a fluorescence microplate reader. A decreased red/green fluorescence intensity ratio indicates brain mitochondrial membrane depolarization [8].
Brain mitochondrial swelling determination
In order to study brain mitochondrial swelling, the isolated mitochondria (0.4 mg/ml) were suspended in respiration buffer containing 150 mM KCl, 5 mM HEPES, 5 mM K2HPO4.3H2O, 5 mM L-glutamate, and 5 mM Pyruvate sodium salt, and the absorbance of mitochondrial suspension was measured at 540 nm using a microplate reader. A reduction in absorbance at min 0 indicates brain mitochondrial swelling [8].
Protein expression analysis by western blotting
For protein extraction, the brain was lysed in an extraction buffer containing 100 mM NaCl, 25 mM EDTA, 10 mM Tris, 1% v/v Triton X-100, and 1% v/v NP-40, 1X protease inhibitor. The brain was homogenized at 600 rpm using a homogenizer (IKA, Germany). The homogenate was centrifuged at 13000 rpm for 10 min, the supernatant was collected, and the protein concentration was determined. 2 mg/ml of protein was separated by gel electrophoresis on a 10% SDS–polyacrylamide gel, and the protein was transferred onto nitrocellulose membranes. The membranes were incubated in 5% in Tris-buffered saline containing 0.1% Tween 20 with non-fat dry milk or bovine serum albumin. Then, membranes were probed with the primary antibodies including mitochondrial fission proteins: p-Drp1ser616 (1:1000 dilution, Cell signaling technology), and Drp1 (1:1000 dilution, Cell signaling technology); mitochondrial fusion proteins: Mfn1 (1:1000 dilution, Abcam), Mfn2 (1:1000 dilution, Cell signaling technology), and OPA1 (1:1000 dilution, Cell signaling technology); BBB protein: Claudin5 (1:1000 dilution, Cell signaling technology); Apoptotic proteins: Bax (1:1000 dilution, Cell signaling technology), Bcl2 (1:1000 dilution, Abcam), and Pro- and cleaved caspase-3 (1:1000 dilution, Cell signaling technology); AD related proteins: Aβ (1:200 dilution, Santa Cruz biotechnology), AβPP (1:1000 dilution, Cell signaling technology), p-TauThr181 (1:1000 dilution, Cell signaling technology), Tau (1:1000 dilution, Cell signaling technology), p-GSK3βser9 (1:1000 dilution, Cell signaling technology), and GSK3β (1:1000 dilution, Cell signaling technology); Antioxidant protein: SOD2 (1:1000 dilution, Cell signaling technology). GAPDH (1:10000 dilution, Abcam) was used as a house keeping protein. Membrane incubation was done with a horseradish peroxidase-conjugated secondary antibody. The blots were then visualized with an enhanced chemiluminescence (ECL) reagent, and the western blot pictures were taken using a Chemidoc touch imaging system, and the densitrometric analysis was analyzed using ImageJ (NIH image) analysis software [25].
Statistical analysis
All experiments or treatments and data analyses were performed utilizing randomization and blinding. Data were expressed as mean±SEM. Data analyses were done using GraphPad Prism 7.0 software. The difference between groups was tasted by one-way-ANOVA followed by an LSD post hoc test. p < 0.05 was considered a statistically significant.
RESULTS
Cardiac I/R injury disrupted brain mitochondrial dynamics balance, and M1 increased brain mitochondrial fusion after cardiac I/R injury
In this study, the alteration of brain mitochondrial dynamics was determined after cardiac I/R injury. Our western blot data showed that cardiac I/R injury did not alter p-Drp1ser616/Drp1 protein level, in comparison with the sham-operated group (Fig. 2A). In contrast, cardiac I/R injury decreased levels of Mfn1, Mfn2, and OPA1 (Fig. 2B, C). The findings suggested that cardiac I/R injury reduced brain mitochondrial fusion following cardiac I/R injury. Therefore, mitochondrial fusion may play an important role in the regulation of brain pathology in rats with cardiac I/R. Then, we pretreated the rats with the mitochondrial fusion promoter M1 prior to cardiac I/R, and we found that M1 significantly increased Mfn2 protein expression (Fig. 2C), without having any effects on p-Drp1ser616/Drp1, Mfn1, and OPA1, in comparison with rats under cardiac I/R with vehicle treatment (Fig. 2A, B, D). These findings suggested that pretreatment with mitochondrial fusion promoter modulated mitochondrial dynamics by targeting Mfn2 protein to increase mitochondrial fusion in the brain of rats with cardiac I/R injury.
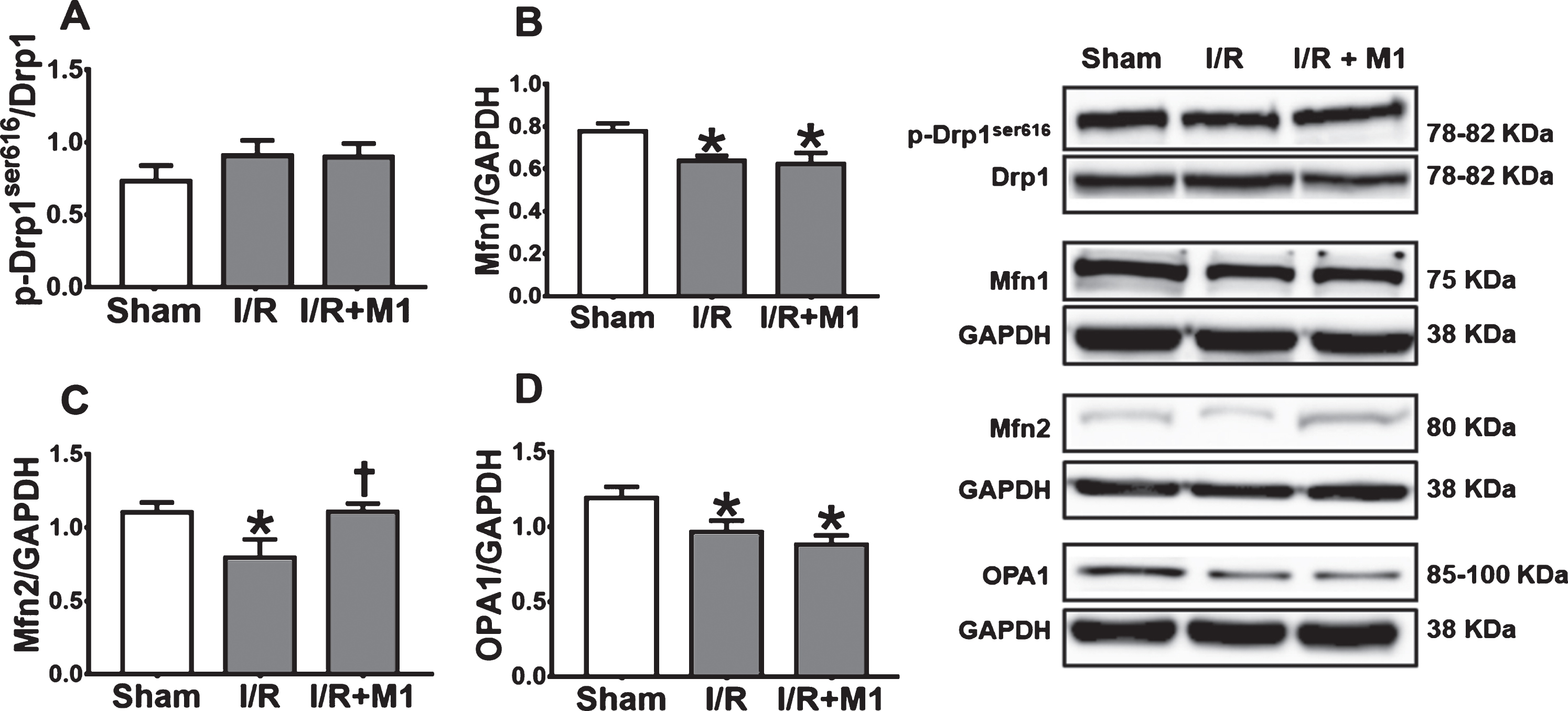
The alterations of brain mitochondrial dynamics in rats with cardiac I/R and the effects of M1 on brain mitochondrial dynamics proteins levels in rats with cardiac I/R. A) p-Drp1ser616/Drp1, B) Mfn1/GAPDH, C) Mfn2/GAPDH, D) OPA1/GAPDH. N = 8 per group. M1, mitochondrial fusion promoter; I/R, ischemia/reperfusion; Drp1, dynamin related protein 1; Mfn1, mitofusin 1; Mfn2, mitofusin 2; OPA1, optic atrophy protein 1. *p < 0.05 versus sham operation group, †p < 0.05 versus cardiac I/R rats treated with vehicle.
Mitochondrial fusion promoter increased BBB tight junction protein, and reduced macrophage infiltration in the brain of rats following cardiac I/R injury
Cardiac I/R injury did not alter % CD11b+/ CD45+low; however, % CD11b+/CD45+high was increased, in comparison with the sham-operated group (Fig. 3A–C). We also found that the level of claudin 5 protein was decreased in the cardiac I/R group, when compared with the sham-operated group (Fig. 3D). Pretreatment with M1 significantly reduced % CD11b+/CD45+high, but it did not affect % CD11b+/CD45+low in rats with cardiac I/R, in comparison with cardiac I/R rats receiving vehicle (Fig. 3A–C). Moreover, pretreatment with M1 significantly increased the level of claudin 5 protein, in comparison with cardiac I/R rats receiving vehicle (Fig. 3D). These findings suggested that cardiac I/R injury led to BBB breakdown, and the macrophages migrated into the brain. Pretreatment with mitochondrial fusion promoter prevented BBB breakdown, and decreased the migration of macrophages to the brain in rats with cardiac I/R injury.

The effects of M1 on microglial function and BBB protein level in rats with cardiac I/R. A) % cells with CD11b+/CD45+low, B) % cells with CD11b+/CD45+high, C) representative pictures of resident and active microglia using a flow cytometry, D) Claudin5/GAPDH. N = 8 per group. M1, mitochondrial fusion promoter; I/R, ischemia/reperfusion; CD, cluster differentiation. *p < 0.05 versus sham operation group, †p < 0.05 versus cardiac I/R rats treated with vehicle.
Mitochondrial fusion promoter reduced brain mitochondrial dysfunction and apoptosis, but it did not reduce mitochondrial oxidative stress in rats following cardiac I/R injury
Brain mitochondrial dysfunction occurred after cardiac I/R injury, indicated by increasing of brain mitochondrial swelling, brain mitochondrial membrane depolarization, and brain mitochondrial ROS production when compared with sham-operated group (Fig. 4A–C). Pretreatment with M1 effectively restored brain mitochondrial membrane potential and reduced brain mitochondrial swelling in rats with cardiac I/R, in comparison with cardiac I/R rats receiving vehicle (Fig. 4B, C). Although M1 restored brain mitochondrial membrane potential and reduced mitochondrial swelling, it could not reduce excessive ROS production in the brain mitochondria following cardiac I/R (Fig. 4A). These findings suggested that cardiac I/R led to brain mitochondrial dysfunction, and pretreatment with mitochondrial fusion promoter improved brain mitochondrial function, but it could not reduce mitochondrial oxidative stress level in brain of rats following cardiac I/R injury. Furthermore, the levels of SOD2 were increased in the brain after cardiac I/R; however, pretreatment with M1 did not affect the levels of SOD2 (Fig. 4D). This data suggested that cardiac I/R increased brain antioxidant levels, which could be a compensatory mechanism against brain injury. Pretreatment with M1 failed to further enhance antioxidant levels, compared to the vehicle group. These data confirm that M1 did not affect the oxidant-antioxidant system in the brain after cardiac I/R.
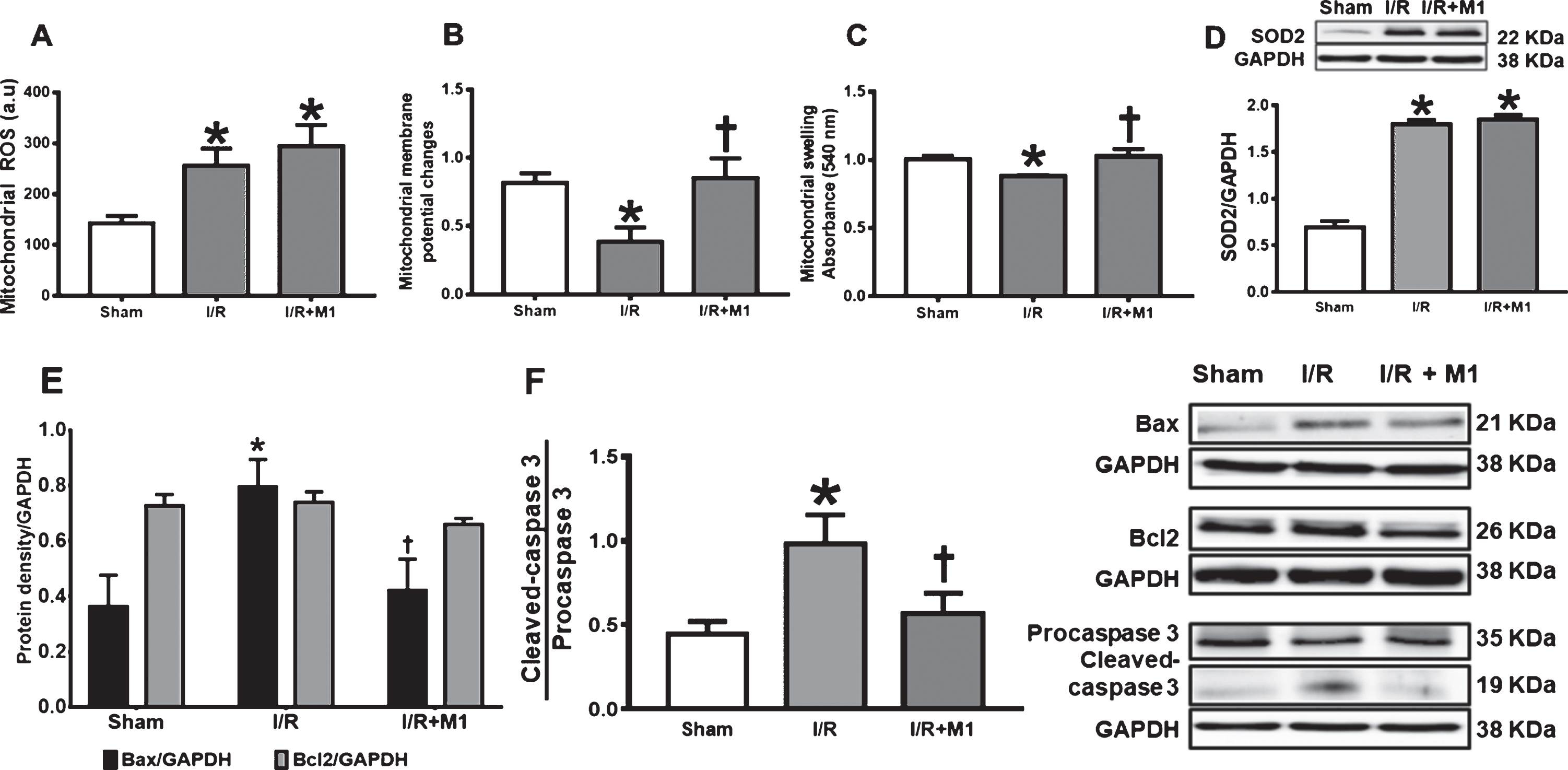
The effects of M1 on brain mitochondrial function and apoptosis in rats with cardic I/R. A) Brain mitochondrial ROS level, B) brain mitochondrial membrane potential changes, C) brain mitochondrial swelling, D) SOD2/GAPDH, E) Bax/GAPDH and Bcl2/GAPDH, F) cleaved-caspase 3/procaspase 3 ratio. N = 8 per group. M1, mitochondrial fusion promoter; I/R, ischemia/reperfusion; ROS, reactive oxygen species; Bax, Bcl-2-associated X protein; Bcl2, B-cell lymphoma 2. *p < 0.05 versus sham operation group, †p < 0.05 versus cardiac I/R rats treated with vehicle.
Brain mitochondrial dysfunction is associated with apoptosis, and we found that cardiac I/R injury increased Bax and cleaved caspase-3/procaspase-3 ratio, which is a pro-apoptotic protein, in comparison with sham-operated group (Fig. 4E, F). However, cardiac I/R did not alter level of Bcl2 protein, which is an anti-apoptotic protein, when compared with sham-operated group (Fig. 4E). Pretreatment with M1 reduced levels of Bax and cleaved caspase-3/procaspase-3 ratio, without effect on level of Bcl2 protein, in comparison with cardiac I/R rats treated with vehicle (Fig. 4E,F). These findings suggested that cardiac I/R caused brain apoptosis, and pretreatment with mitochondrial fusion promoter decreased the susceptibility of brain to apoptosis in rats following cardiac I/R injury.
Mitochondrial fusion promoter reduced the expression of AD-related proteins in rats with cardiac I/R injury
Cardiac I/R injury increased levels of brain Aβ, p-Tau, and tau proteins, but it did not affect AβPP protein level, in comparison with sham-operated group (Fig. 5A–D). Pretreatment with M1 significantly reduced levels of brain p-Tau, and tau proteins in rats following cardiac I/R, in comparison with cardiac I/R rats receiving vehicle (Fig. 5B–D). The results showed that the ratio of p-Tau/tau was no different between groups (Fig. 5D). However, M1 did not affect the level of brain Aβ and AβPP proteins in rats following cardiac I/R, in comparison with cardiac I/R rats receiving vehicle (Fig. 5A). These finding suggested that cardiac I/R led to accumulation of AD-related proteins, and pretreatment with mitochondrial fusion promoter attenuated AD pathology by decreasing hyperphosphorylated tau, but it did not alter Aβ in rats following cardiac I/R injury. Glycogen synthase kinase 3β (GSK3β) phosphorylates tau in serine and threonine residues. It also contributes to Aβ formation [26]. Therefore, we performed an experiment to determine the effects of M1 on the protein expression of p-GSK3β/GSK3β. Our results demonstrated that the ratio of p-GSK3β/GSK3β was increased in the brain following cardiac injury, in comparison with sham-operated group. Pretreatment with M1 significantly reduced p-GSK3β/GSK3β (Fig. 5E).

The effects of M1 on Alzheimer’s disease related proteins in rat brains with cardiac I/R. A) Amyloid-β/GAPDH, B) AβPP/GAPDH, C) p-Tau/GAPDH and tau/GAPDH, D) p-Tau/tau ratio, E) p-GSK3βSer9/GSK3β ratio, N = 8 per group. M1, mitochondrial fusion promoter; I/R, ischemia/reperfusion; AβPP, amyloid-β protein precursor. *p < 0.05 versus sham operation group, †p < 0.05 versus cardiac I/R rats treated with vehicle.
DISCUSSION
The significant findings from this study are: 1) cardiac I/R injury caused imbalanced brain mitochondrial dynamics by reducing mitochondrial fusion; 2) Pretreatment with mitochondrial fusion promoter enhanced level of Mfn2 protein in the brain following cardiac I/R injury; 3) Pretreatment with this mitochondrial fusion promoter reduced BBB breakdown, resulting in reduced macrophage infiltration and brain inflammation in rats following cardiac I/R injury; and 4) Pretreatment with mitochondrial fusion promoter reduced brain mitochondrial dysfunction, apoptosis, and hyperphosphorylated tau, as indicated by decreased p-Tau and p-GSK3β in rats following cardiac I/R injury.
Previous studies demonstrated that cardiac I/R injury caused [13] deleterious effects in many organs, including the brain, by inducing oxidative stress and inflammation [6, 27]. The reduction in transient cerebral blood flow following cardiac I/R injury, indicated that brain dysfunction had occurred [13]. Brain damage following cardiac I/R resulted from BBB breakdown, oxidative stress, mitochondrial dysfunction, brain apoptosis, and accumulation of AD-related proteins [6, 11]. This study indicates that treatment by a mitochondrial fusion promoter might be another possible approach for patients with AD with a previous history of heart attack because of reduction of hyperphosphorylated tau after treatment. On the other hand, Aβ was not reduced after pretreatment with the mitochondrial fusion promoter. This finding could be due to a short treatment duration, or the fact that the mitochondrial fusion promoter was given to the rats as a single dose. There was no evidence to show that M1 can cross the BBB, even though the pretreatment with M1 could cause beneficial effects on BBB breakdown as presented by a decrease in claudin 5 protein expression after M1 treatment. Thus, further studies involving different doses and duration or extension of the administration of M1 in cardiac I/R injury are needed to clarify the findings. Following cardiac I/R injury, an imbalance in brain mitochondrial dynamics was acknowledged as one of the pathogenic factors that leads to the associated brain damage [7, 28]. In order to confirm that the imbalance of mitochondrial dynamics was the major cause of the brain disorders following the cardiac I/R injury, a brain mitochondrial dynamics analysis was done in this study. Our findings demonstrated that cardiac I/R injury precipitated the reduction of the mitochondrial fusion related proteins (Mfn1, Mfn2, and OPA1) without the alterations in mitochondrial fission related proteins (p-Drp1ser616, Drp1). Thus, the results implied that brain mitochondrial fusion is the important target for the maintenance of mitochondrial homeostasis and cellular function in the brain during cardiac I/R injury.
The efficacy of a mitochondrial dynamics modulator has been reported in animals with cardiac I/R injury and cerebral I/R injury [19, 20]. According to our results, only mitochondrial fusion was decreased in the brain after cardiac I/R, therefore, the mitochondrial fusion promoter, M1, was used in this study. The beneficial effects of M1 were observed in the heart during the process of cardiac I/R injury. It could lead to a reduction in myocardial infarct size, arrhythmias, and cardiac dysfunction by improving the regulation and function of cardiac mitochondrial dynamics [20]. This is the first study to demonstrate the protective effects of M1 on the brain during cardiac I/R injury.
M1 is a mitochondrial fusion promoter which effectively induces mitochondrial elongation by supporting basal mitochondrial activity (through Mfn1 and Mfn2) at the outer mitochondrial membrane [29]. Although pharmacological interventions using M1 or other mitochondrial fusion promoters have been used to restore the mitochondrial dynamic balance and reverse the brain damage [30], M1 only effectively restored Mfn2 protein expression with no effects on Mfn1 and OPA1 in the brain of rats following cardiac I/R injury. Enhancing Mfn2 expression could reduce brain mitochondrial dysfunction as indicated by decreasing brain mitochondrial membrane depolarization and mitochondrial swelling. However, enhancing mitochondrial fusion could not reduce brain mitochondrial oxidative stress after cardiac I/R. These data demonstrated that an improvement in brain mitochondrial function in M1 therapy is independent to level of oxidative stress. Nevertheless, our data contradicted with the previous in vitro study, they suggested that M1 significantly reduced mitochondrial ROS levels in the Aβ-treated primary hippocampal neurons [28]. This discrepancy might be due to a different in brain model used in the study. Hippocampus is more vulnerable to stress than in the cortex [31]. This might be a reason that why we could not observe the beneficial effects of M1 in reducing mitochondrial ROS levels in our brain mitochondria, which isolated from the whole brain. The effects of M1 on mitochondrial ROS levels in the hippocampus of rats following cardiac I/R should be investigated in future study.
In addition to oxidative stress, brain inflammation has been proposed as another mechanism responsible for brain damage after cardiac I/R [6]. The mitochondrial fusion promoter effectively reduced macrophage infiltration via increasing BBB tight junction proteins, which led to a reduction in brain inflammation. Previous studies suggested that brain inflammation could led to an activation and accumulation of AD proteins [6]. A reduction in brain inflammation by a mitochondrial fusion promoter might be another possible mechanism in causing the reduction in hyperphosphorylated tau. Although tau protein is reportedly phosphorylated at thr181 residue in ischemic stroke [32] and our data revealed that M1 pretreatment could attenuate this phosphorylation, the effects of M1 on other phosphorylated residues of tau should also be determined in the future study. On the other hand, Aβ was not reduced after pretreatment with the mitochondrial fusion promoter. This finding could be due to a short treatment duration, and mitochondrial fusion promoter being given to the rats as a single dose. An association between brain mitochondrial function and BBB permeability has been reported [33]. The authors of that study suggested that reductions of oxidative phosphorylation and electron transport chain subunits increased BBB permeability and disrupted cell to cell tight junctions in mice with transient ischemic stroke [33]. A crucial role of Mfn2 was demonstrated in a previous study which used 3T3-L1 fibroblast cells. Mfn2 knockdown reduced oxidative phosphorylation and mitochondrial metabolism [34].
Both brain mitochondrial dysfunction and brain inflammation are key initiators of apoptosis [35]. Cardiac I/R injury causes both brain mitochondrial dysfunction and brain inflammation, which could induce apoptosis. Our data showed that Bax and cleaved caspase 3 expression were increased after cardiac I/R, and it could be suppressed by a pretreatment with the mitochondrial fusion promoter. These data suggested that the mitochondrial fusion promoter decreased the susceptibility of the brain to apoptosis in rats with cardiac I/R injury through reduction in brain mitochondrial dysfunction and brain inflammation.
Conclusions
Our findings suggest that cardiac I/R injury causes brain damage and impaired brain mitochondrial balance. Pretreatment with mitochondrial fusion promoter effectively increases Mfn2 expression, led to reduction in BBB breakdown, brain inflammation, and brain mitochondrial dysfunction, resulting in reduced brain apoptosis in rats with cardiac I/R. Therefore, the mitochondrial promoter (M1) may be a candidate for future alternative treatment in order to restore mitochondrial homeostasis and reduce the deleterious effects in the brain after cardiac I/R injury.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Senior Research Scholar grant from the National Research Council of Thailand (SCC); a NSTDA Research Chair Grant from the National Science and Technology Development Agency Thailand (NC), and a Chiang Mai University Center of Excellence Award (NC).