
Abstract
Background:
The recent developed PET ligands for amyloid-β (Aβ) and tau allow these two neuropathological hallmarks of Alzheimer’s disease (AD) to be mapped and quantified in vivo and to be examined in relation to cognition.
Objective:
To assess the associations among Aβ, tau, and cognition in non-demented subjects.
Methods:
Three hundred eighty-nine elderly participants without dementia from the Alzheimer’s Disease Neuroimaging Initiative underwent tau and amyloid PET scans. Cross-sectional comparisons and longitudinal analyses were used to evaluate the relationship between Aβ and tau accumulation. The correlations between biomarkers of both pathologies and performance in memory and executive function were measured.
Results:
Increased amyloid-PET retention was associated with greater tau-PET retention in widespread cortices. We observed a significant tau increase in the temporal composite regions of interest over 24 months in Aβ+ but not Aβ– subjects. Finally, tau-PET retention but not amyloid-PET retention significantly explained the variance in memory and executive function. Higher level of tau was associated with greater longitudinal memory decline.
Conclusion:
These findings suggested PET-detectable Aβ plaque pathology may be a necessary antecedent for tau-PET signal elevation. Greater tau-PET retention may demonstrate poorer cognition and predict prospective memory decline in non-demented subjects.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by the pathological deposition of both amyloid-β (Aβ) and tau. In the research framework proposed by the National Institute on Aging-Alzheimer’s Association (NIA-AA) workgroup, Alzheimer’s pathologic change refers to the early stage of the Alzheimer’s continuum, defined in vivo by biomarkers as abnormal Aβ deposition but normal tau status. The term “Alzheimer’s disease” would be applied when biomarker evidence of both Aβ and pathologic tau was present [1]. The Aβ cascade hypothesis proposes that amyloidosis induces the spread of pathologic tau and that pathological tau subsequently leads to neurodegeneration which eventually causes cognitive decline [1–4].
Recently, the developed positron emission tomography (PET) ligands for Aβ [5, 6] and tau [7] permit the investigation of these two neuropathological hallmarks of AD in vivo. Previous PET imaging studies showed that in clinically normal subjects with a negative amyloid-PET scan, tau-PET retention was limited largely to medial temporal lobe [8], whereas in subjects with positive amyloid PET scans, cross-sectional comparisons suggested increasing and progressive tau-PET retention in widespread cortices [8].
Despite the growing cross-sectional tau-PET imaging literature, only a few studies examined longitudinal change in tau-PET signal. Previous longitudinal studies indicated that the rate of tau retention associated with baseline tau retention and Aβ status measured by amyloid-PET [8–10]. Further evaluation of the way in which tau changes over time could be critical to understand the interaction between amyloid and tau in promoting neurodegeneration and cognitive impairment in AD [8].
Cortical composite and regional tau-PET standard uptake value ratio (SUVr) has been reported to correlate with cognitive impairment [11–16], but the relationship between tau-PET SUVr and specific cognitive domains varies across studies [15–19]. Besides, inconsistent results were found on how Aβ and tau correlated with cognition. While some studies concluded that only tau tangles correlated with cognition and clinical symptoms [13, 20], others showed that Aβ and tau were synergistically associated with cognitive decline [20].
Here, we hypothesize that increased amyloid level can predict increased tau-PET deposit in certain regions of interest (ROI), and increased tau is associated with memory and executive function decline. We stratified a large cohort of non-demented elderly participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) into two groups by Aβ status using florbetapir PET imaging. The differences in baseline retention and prospective change of tau pathology as measured by [F-18]-AV-1451 PET between groups was investigated. We then evaluated the associations of baseline tau burden and baseline Aβ burden with the rate of prospective tau accumulation. Finally, we analyzed the relative contributions of Aβ, tau, and the potential synergy of these pathologies in the prediction of memory and executive function performance and longitudinal decline.
MATERIALS AND METHODS
Study subjects and ADNI database
We obtained the data used in this article from the Alzheimer’s Disease Neuroimaging Initiative-3 (ADNI-3) database (http://adni.loni.usc.edu). ADNI is a longitudinal multi-center natural history study designed to characterize clinical imaging, genetic, and biochemical biomarkers for early detection and tracking of AD. In 2016, ADNI-3 was initiated with an expanded goal of determining the relationships between the clinical, cognitive, imaging, genetic, and biochemical biomarker characteristics across the entire spectrum of AD, in which 1,070–2,000 participants were enrolled across cognitively normal (CN), mild cognitive impairment (MCI), and AD dementia cohorts in ADNI-3. Detailed inclusion and exclusion criteria are presented in the database. Importantly, ADNI-3 added brain scans that detect tau protein tangles (tau-PET), a key indicator of the disease (http://adni.loni.usc.edu/about). This study was approved by institutional review boards of all contributing research institutions, and informed consent in writing was acquired from all subjects or authorized agents (http://adni.loni.usc.edu/methods/documents).
We recruited three hundred eighty-nine non-demented subjects from ADNI-3 who had both amyloid and tau PET scans at baseline. We chose 2 years as the time window for longitudinal analyses [8, 10]. Tau-PET scans were performed for 104 participants at month 12 and for 68 at month 24. Meanwhile, 18 and 56 participants received amyloid-PET scans at month 12 and month 24, respectively. Either CN or MCI participants were included in this present study. The diagnostic criteria for CN and MCI in ADNI were previously described [21]. Diagnostic grouping was based on the most recent change in clinical diagnosis evaluation provided by the ADNI Clinical Core. Participants were also classified by Aβ elevated status as amyloid positivity (Aβ+) or negative (Aβ–) using amyloid-PET global cortical SUVr.
Imaging acquisition and analysis
PET imaging was performed at each ADNI site according to standardized protocols, which was published previously [20]. Data of tau PET imaging assessed by [F-18]-AV-1451 and of amyloid PET imaging assessed by florbetapir ([F-18]-AV-45) were pre-processed by UC Berkeley and Lawrence Berkeley National Laboratory.
In the present study, the amyloid-PET cortical summary values were computed as the average of the mean SUVr in the four main cortical regions (frontal, anterior/posterior cingulate, lateral parietal, and lateral temporal) using the amyloid-PET mean and volume of each sub-regions. The amyloid-PET SUVR was normalized by the whole cerebellum reference region [22]. Amyloid positivity was defined as amyloid-PET cortical summary SUVr > 1.11 based on the University of California, Berkeley quantification [6, 23]. This cutoff was equivalent to the upper 95% confidence interval above the mean of a group of young normal controls, and was recommended by ADNI [24]. The tau-PET SUVr was calculated based on mean tau-PET uptake normalized to uptake in a grey matter masked cerebellum reference region as previously described [20]. We calculated a previously reported temporal composite SUVr to capture the characteristics of cross-sectional and longitudinal tau-PET. The temporal composite was calculated as the voxel-number weighted average of tau-PET SUVr in the entorhinal cortex, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal regions of interest [10, 25]. This meta-region of interest was defined in a previous study with a cross-sectional design to separate cognitively unimpaired subjects with Aβ+ and cognitive impaired subjects with Aβ– [25]. Another longitudinal tau-PET study showed this temporal composite could effectively capture the annual change of tau-PET SUVr [10]. Partial volume correction (PVC) was not applied to Tau-PET and amyloid-PET data.
Cognitive assessment
All subjects underwent cognitive assessment based on Mini-Mental State Examination (MMSE) score, Alzheimer’s Disease Assessment Scale, 11- Item Subscale (ADAS11) score, and composite scores on two cognitive domains, namely memory and executive function. The composite score for memory was composed of scores of the Rey’s Auditory Vocabulary List Test (RAVLT), ADAS11, Logical Memory (LM), and MMSE recall scores [26]. The composite score for executive function included Category Fluency (animals and vegetables scores), Trail Making Test (TMT) A and B, Digit span backwards, Wechsler Adult Intelligence Scale-Revised (WAIS-R) Digit Symbol Substitution, and five Clock drawing items (circle, symbol, numbers, hands, time) [20, 27]. These tests were administered as previously described (http://adni.loni.usc.edu/methos/documents).
Statistical analysis
Baseline characteristics were compared between Aβ+ and Aβ– subjects using 2-tailed Wilcoxon rank-sum tests for continuous variables and χ2 tests for categorical variables.
We used analyses of covariance to evaluate differences in regional and temporal composite tau-PET SUVr across clinical diagnosis groups and Aβ status, adjusting for baseline age, gender, educational years, and APOE ɛ4 carriage. Planned contrasts within the model evaluated differences among CN and MCI subjects by Aβ status (i.e., within the Aβ+, or Aβ– subgroups). Similarly, the tau-PET SUVr were also compared within each diagnostic group (i.e., CN, or MCI subgroups) according to Aβ status through planned contrasts [14]. Log transformation was applied to tau-PET SUVr to fulfill normality assumptions.
Mixed effect model was fitted to evaluate the annual tau change in temporal composite ROI with covariates including baseline temporal composite tau-PET SUVr, age, gender, educational years, and APOE ɛ4 carriage in Aβ+ and Aβ- groups separately [8]. We subsequently examined baseline cortical Aβ (continuous) and baseline temporal composite tau as predictors of the rate of regional tau accumulation in separate models, adjusting for age, gender, educational years, and APOE ɛ4 carriage. For an exploratory analysis, we also examined baseline temporal composite tau as predictor of the annualized change of cortical Aβ, adjusting for age, gender, educational years, and APOE ɛ4 carriage.
We assessed the correlations between biomarkers of both pathologies and cognition with multiple linear regression models. Baseline memory and executive function composite scores were used as dependent variables, with baseline cortical amyloid-PET retention (continuous) and baseline temporal composite tau-PET retention as simultaneous predictors. Age, gender, educational years, and APOE ɛ4 carriage were regarded as confounding variables. Then we added the two-way interaction between baseline cortical Aβ and baseline temporal composite tau to this model to determine whether the association between tau and cognitive impairment was modified by Aβ levels [16]. We then examined baseline cortical Aβ and baseline temporal composite tau as simultaneous predictors of longitudinal memory and executive function change in the same mixed effect model, and we subsequently added the three-way interaction between Aβ, tau and time to this model.
Statistical significance was defined as p < 0.05 for all analyses. Statistical analyses were performed using the R statistical software (version 3.6.1) and SPSS Statistics 23.
RESULTS
Table 1 showed the demographics and baseline characteristics for the 389 subjects with available tau-PET and amyloid-PET scans, including 160 Aβ+ subjects and 229 Aβ– subjects. Aβ+ subjects were older (p < 0.001); were more likely to be APOE ɛ4 carriers (p < 0.001); and had lower MMSE score (p < 0.001), higher ADAS11 score (p = 0.001), lower composite memory score (p < 0.001), and lower composite executive function score (p < 0.001). There were no significant differences in educational years and gender composition between Aβ+ and Aβ– subjects. Demographics were also presented by clinical diagnostic groups (Supplementary Table 1).
Characteristics of the Participants1
1Data were mean (SD) or number (%) unless otherwise stated. Participants with amyloid-PET cortical summary SUVr > 1.11 were classified as amyloid-β positive; those with amyloid-PET cortical summary SUVr≤1.11 were classified as the amyloid-β negative. n, number; Aβ+, amyloid-β positive; Aβ–, amyloid-β negative; MMSE, Mini-Mental State Examination; ADAS11, Alzheimer’s Disease Assessment Scale, 11-Item Subscale; Mem, memory; EF, executive function.
Table 2 showed the different patterns of cross-sectional tau-PET signal in Aβ+ and Aβ– subjects. In Aβ+ subjects, the temporal composite and other regional tau-PET SUVr was elevated for MCI compared with the CN (Temporal composite: p < 0.001; Frontal: p = 0.002; Occipital: p = 0.02; Parietal: p = 0.002; Temporal: p < 0.001). Within the Aβ- group, the differences in tau-PET SUVr between CN and MCI were detected in temporal composite ROI (p = 0.031), frontal lobe (p = 0.014), and temporal lobe (p = 0.041). Within CN and MCI group, the tau-PET SUVr of Aβ+ subjects significantly elevated in the temporal composite (CN: p < 0.001; MCI: p < 0.001), frontal lobe (CN: p = 0.003; MCI: p = 0.003), parietal lobe (CN: p = 0.002; MCI: p = 0.001), occipital lobe (CN: p = 0.223; MCI: p = 0.007), and temporal lobe (CN: p < 0.001; MCI: p < 0.001). As shown in Fig. 1, temporal composite tau-PET SUVr was higher for MCI group compared to the CN group and for the Aβ+ compared to the Aβ–.
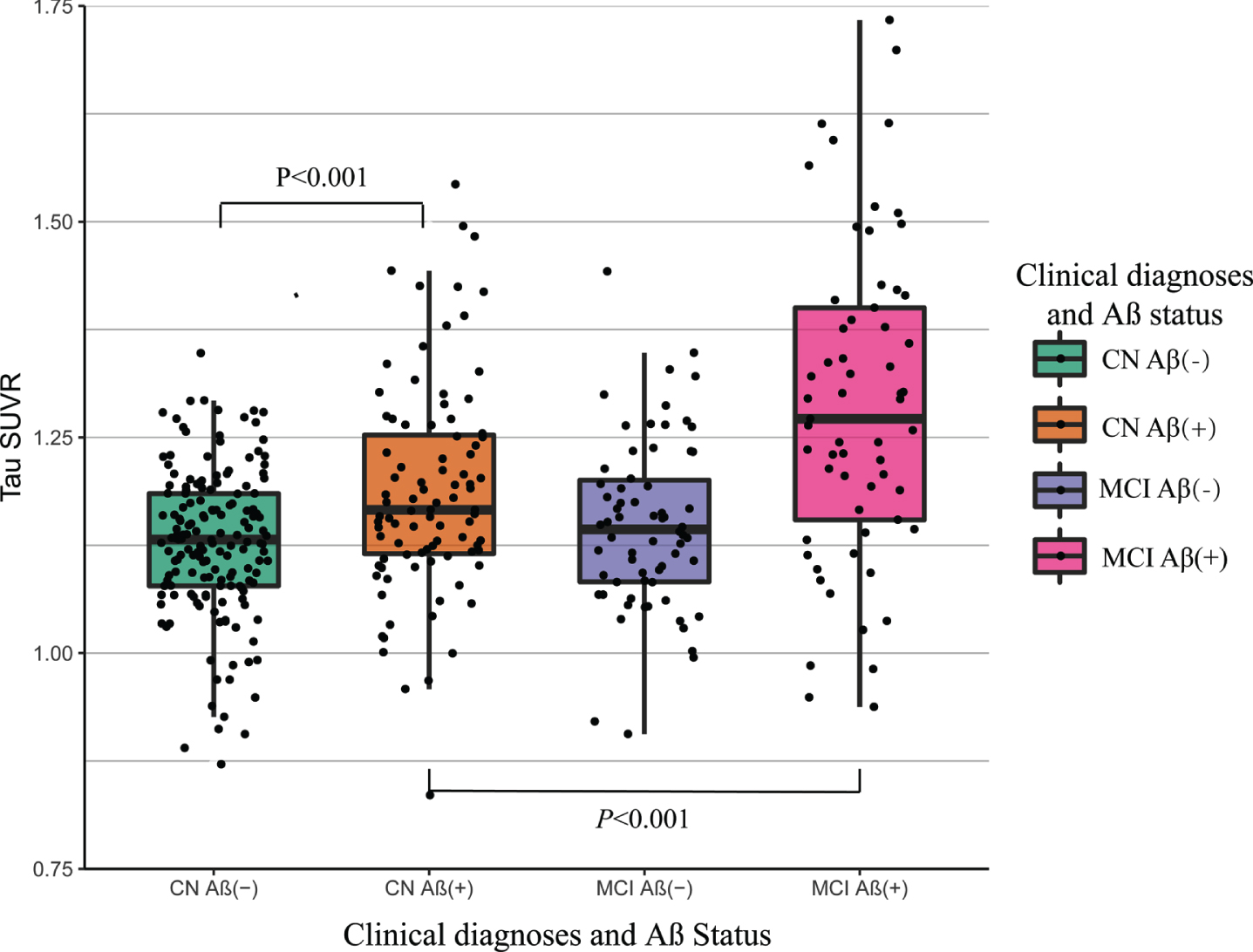
Temporal composite tau-PET SUVr by clinical diagnostic cohort and amyloid status. Box plot of temporal composite tau-PET SUVr by diagnostic cohort and amyloid status of all 389 participants. Participants with amyloid-PET cortical summary SUVr > 1.11 were classified as amyloid-β positive; those with amyloid-PET cortical summary SUVr≤1.11 were classified as the amyloid-β negative. p values are from the analysis of covariance with planned contrast. The box plot whiskers extend to the lowest and highest data points within 1.5 times the inter quartile range from the lower and upper quartiles. The dots represent individual points. Aβ(+), amyloid-β positive; Aβ(–), amyloid-β negative; CN, cognitively normal, MCI, mild cognitive impairment; SUVr, standard uptake value ratio.
Tau-PET SUVr by clinical diagnostic group and amyloid status1
1Data were shown in mean (SD) or number unless otherwise stated. Participants with amyloid-PET cortical summary SUVr > 1.11 were classified as amyloid-β positive; those with amyloid-PET cortical summary SUVr≤1.11 were classified as the amyloid-β negative. Comparison were adjusted for baseline age, gender, educational years and APOE ɛ4 carriage. Log transformation was applied to tau-PET SUVr to fulfill normality assumptions. n, number; Aβ+, amyloid-β positive; Aβ–, amyloid-β negative; CN, cognitively normal, MCI, mild cognitive impairment; SUVr, standard uptake value ratio.
Figure 2 showed the significant longitudinal change of temporal composite tau-PET SUVr over 24 months in Aβ+ but not Aβ– subjects. The baseline temporal composite tau retention was significantly higher in the Aβ+ subjects compared to Aβ– subjects [1.25(0.120) versus 1.14(0.083), between-group difference p < 0.001]. In the Aβ+ group, the tau-PET SUVr increased significantly [Estimated annualized change (95% CI)=0.01(0.004∼0.018), p = 0.003]. In the Aβ– group, the tau-PET SUVr remained unchanged over time [Estimated annualized change (95% CI)=0.00(–0.009∼0.006), p = 0.722].
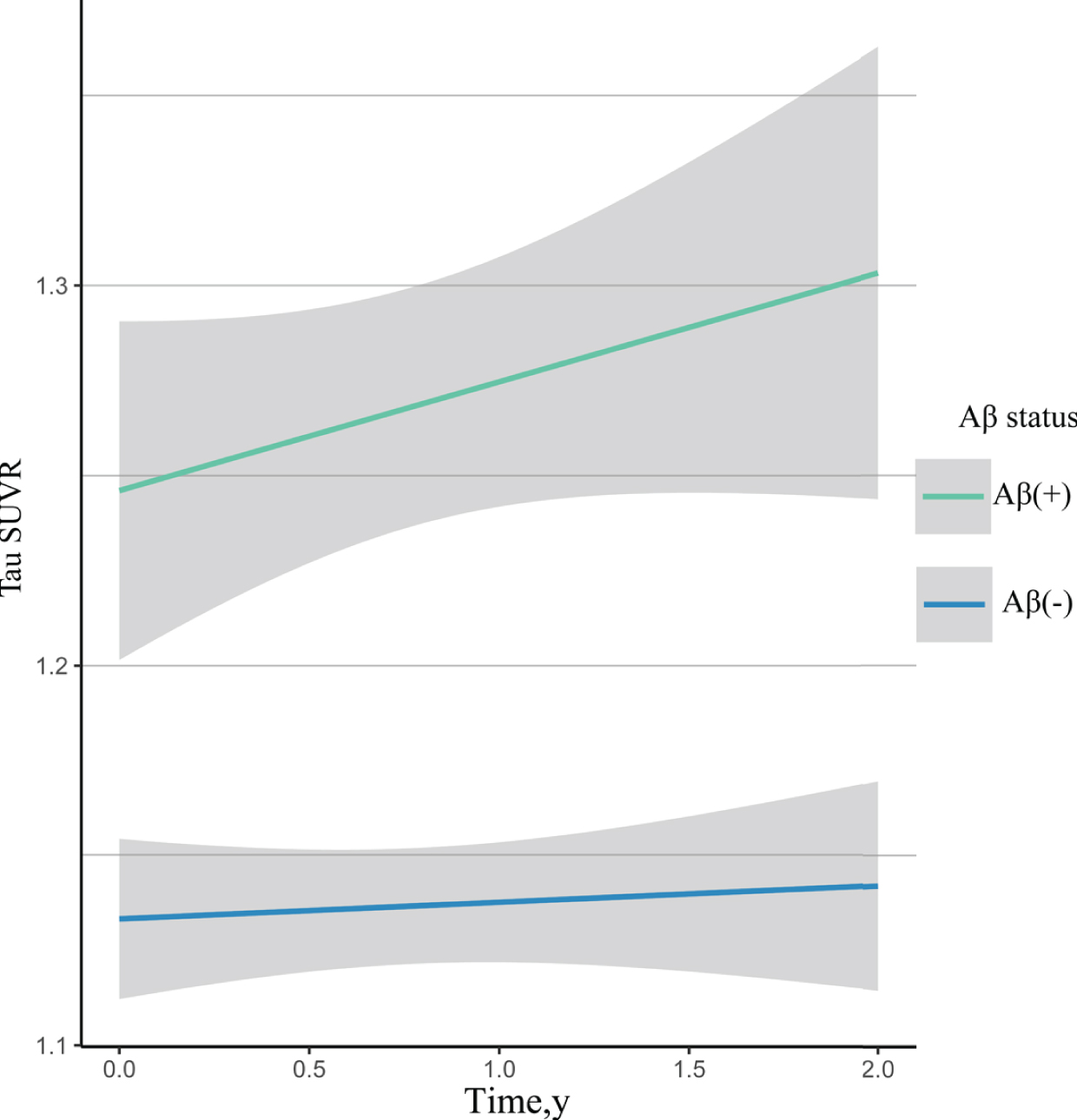
Estimated temporal composite tau-PET SUVr annualized change. Participants with amyloid-PET cortical summary SUVr > 1.11 were classified as amyloid-β positive; those with amyloid-PET cortical summary SUVr≤1.11 were classified as the amyloid-β negative. Aβ(+), amyloid-β positive; Aβ(–), amyloid-β negative; SUVr, standard uptake value ratio.
As shown in Table 3, baseline cortical Aβ burden and baseline temporal composite tau burden were associated with longitudinal regional tau accumulation. Higher levels of Aβ were associated with greater longitudinal tau change in temporal composite ROI [β= 0.05±0.013, p < 0.001], occipital lobe [β= 0.04±0.012, p = 0.005], parietal lobe [β= 0.04±0.012, p = 0.005], and temporal lobe [β= 0.04±0.012, p = 0.001]. Greater baseline tau in temporal composite ROI also related to greater tau accumulation in temporal composite ROI [β= 0.05±0.015, p < 0.001], occipital lobe [β= 0.03±0.014, p = 0.020], parietal lobe [β= 0.04±0.014, p = 0.008], and temporal lobe [β= 0.05±0.014, p = 0.001]. The effect of baseline temporal composite tau on annualized Aβ change was additionally investigated and we did not find evidence of a significant relationship between prospective Aβ change and baseline temporal composite tau [β= 0.02±0.022, p = 0.265].
Effects of baseline Tau and baseline cortical Aβ on prospective tau change1
1Data were Estimate (standard errors) unless otherwise stated. All models include covariates for baseline age, gender, educational years and APOE ɛ4 carriage. Log transformation was applied to tau-PET standard uptake value ratio (SUVr) to fulfill normality assumptions. Aβ, amyloid-β; ROI, regions of interest.
The associations of Aβ and tau with cognition were lastly examined (Table 4; Supplementary Table 2). Greater temporal cortical tau at baseline was related to poorer memory [β= –1.32±0.464, p = 0.004] and executive function [β= –1.47±0.604, p = 0.015]. In contrast, the relationships between baseline cortical Aβ and cognitive performances did not reach statistical significance and were considerably smaller in magnitude than those observed for baseline temporal composite tau [memory composite: β= –0.24±0.62, p = 0.693; executive functional composite: β= –0.41±0.807, p = 0.611]. In the longitudinal analyses, baseline temporal composite tau predicted greater longitudinal decline in the memory composite [β= –0.41±0.189, p = 0.033], but baseline cortical Aβ was not associated with the memory change. Neither Aβ nor tau was observed to be associated with prospective change of executive function. The interaction between Aβ and tau did not show significant effect on cross-sectional and longitudinal cognitive performance for composites of memory and executive function, which indicated that the effect of tau on cognition was not modified by Aβ.
Effects of baseline Tau and baseline cortical Aβ on cognitive performance at baseline1
1Data were Estimate (standard errors) unless otherwise stated. All models include covariates for baseline age, gender, educational years and APOE ɛ4 carriage. Aβ, amyloid-β; Mem, memory; EF, executive function.
DISCUSSION
This study investigated the different patterns of cross-sectional tau-PET signal in Aβ+ and Aβ– groups. In the longitudinal analysis, we observed a significant increase in temporal composite ROI over 24 months. This increase was amyloid-dependent, correlated with baseline tau burden. Finally, tau-PET retention significantly explained the variance in memory and executive function composite scores, and this was independent of the amyloid-PET retention. Elevated tau-PET signal could predict prospective memory decline.
According to the research framework proposed by NIA-AA, cognitively unimpaired individuals with normal amyloid PET were not in the Alzheimer’s continuum and cognitively unimpaired individuals with abnormal amyloid PET were early in the Alzheimer’s continuum [1]. We found that elevated tau retention in CN Aβ+ subjects relative to CN Aβ– subjects. This replicated previous reports that Aβ and tau PET measures were correlated, even among CN [16]. Furthermore, this change was not confined to the temporal lobe, suggesting that early tau accumulation may not be as spatially restricted as implied in Braak staging [10, 28]. Taken together, subjects with abnormal amyloid-PET scans may have increasing tau-PET retention in widespread cortices, even at very early stage of AD.
In subjects with positive amyloid-PET scans, cross-sectional comparisons suggested as the disease progresses, pathological tau accumulated within and beyond the temporal lobe. In subjects with negative amyloid-PET scans, the increasing tau retention was limited to temporal lobe and frontal lobe. These findings suggested that Aβ plaques may potentiate the spread of tau beyond the temporal lobe [8]. Previous studies identified a propagation pathway for tau accumulation. Particularly, media/inferior temporal lobe areas projected pathways of tau-propagation toward the anterior pole, lateral and postero-medial temporal cortex, and midline frontal regions such as the orbitofrontal cortex [29]. In the present study, we found the different spatial pattern of tau progression in Aβ+ and Aβ– subjects. This suggested that Aβ might be associated with the tau propagation patterns. Further studies are wanted to investigate of association of Aβ and tau propagation pathway.
Early in the disease, tau-PET SUVr was elevated in the temporal composite ROI (CN Aβ– versus CN Aβ+). This indicated that this ROI could capture the change in tau PET seen early in the Alzheimer’s continuum. While later in the disease, the temporal composite tau-PET SUVr increased as the disease progressed (CN Aβ+ versus MCI Aβ+). These results suggested that rather than measure progression as change from lower to higher Braak stages, progression could also be measured as increasing tau-PET SUVr within this temporal composite ROI. Thus, quantification of change in tau-PET can be accomplished [10].
The absence of measurable change in temporal composite tau in Aβ- subjects, in contrast to the significant change in Aβ+ subjects (Fig. 2), suggested that amyloid deposition may be a necessary antecedent for tau accumulation [4, 9]. Mixed effect model also pointed to baseline cortical Aβ as the predictor of the rate of tau accumulation (Table 3). The amyloid-cascade hypothesis suggests that Aβ accelerates the spread of tau, disrupting function and initiating neurodegeneration in distributed brain networks, resulting in cognitive decline. This hypothetical model remains to be tested with clinical trails with anti-Aβ therapeutic agents [30]. Additionally, we did not find evidence of significant association between Aβ change and baseline tau burden. This may also support the temporal ordering in the above model.
The rate of different regional tau accumulation varied as a function of the baseline composite temporal tau burden (Table 3). This result was consistent with the hypothesis of trans-synaptic spread of tau [31, 32], in which it would be expected that the higher the baseline tau burden, the greater the number of neurons affected. This, in turn, would cause a greater number of at-risk synapses and thus, a greater degree of elevation in tau over the observation period [8]. This finding may also suggest that temporal cortex areas are the main “giver” regions for tau signal and the temporal lobe pathology progresses distantly [29].
We found baseline temporal composite tau was significantly associated with memory and executive function impairment. This was in agreement with the pathology literature, which suggested that tau tangles but not Aβ plaques correlated with cognition [33]. Tau tangles are also associated with neurodegeneration [34–36] and are likely to be part of the chain of events leading to cortical dysfunction and cognitive impairment. We also found higher levels of tau may demonstrate prospective memory decline. Our results raised the possibility that halting tau accumulation would prevent cognitive decline, and trials would benefit from tau-PET to identify participants at risk of cognitive decline [20]. Furthermore, baseline cortical Aβ was not closely associated with memory and executive function decline, and the association between tau and cognitive decline was independent of the amyloid-PET retention. This was consistent with previous work suggesting that tau-PET signal was more proximal to cognitive decline than amyloid-PET signal [20]. However, there were other studies showing both Aβ and tau were required for memory decline during preclinical stages of AD [11, 16].
Our study had some limitations. In this study, we only focused on the tau accumulation in composite regions of interest and did not capture the detailed topographic characteristics of tau change. Thus, further longitudinal tau-PET studies may test the sequential pattern of tau accumulation and its associations with amyloid and cognition. Additionally, our analyses considered data without partial volume correction. Partial volume correction can reduce contamination from regions in which off-target binding is common into neighboring regions of interest. Our inferences of these PET imaging need to be scrutinized further in data with partial volume correction.
Conclusions
Our findings suggested elevated tau-PET biomarker may be dependent on PET-detectable amyloid-β plaque pathology. Baseline cortical Aβ burden could accelerate the tau accumulation. Thus it is possible that anti-Aβ therapeutic agents might prevent further accumulation of tau, which may slow cognitive decline. Moreover, tau-PET retention but not amyloid-PET retention significantly explained the variance in memory and executive function composite scores and demonstrated the prospective memory decline. Thus, changes in tau burden measured by tau-PET imaging might yield a stable, reliable and accurate statements about cognition and disease progression.
Footnotes
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (91849126), Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01) and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (![]() ). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.