
Abstract
Background:
Given that there is no specific drug to treat Alzheimer’s disease, non-pharmacologic interventions in people with subjective cognitive decline (SCD) and amnestic mild cognitive impairment (aMCI) are one of the most important treatment strategies.
Objective:
To clarify the efficacy of blue-green (500 nm) light therapy on sleep, mood, and physiological parameters in patients with SCD and aMCI is an interesting avenue to explore.
Methods:
This is a monocentric, randomized, and controlled trial that will last for 4 weeks. We will recruit 150 individuals aged 45 years or older from memory clinics and divide them into 5 groups: SCD treatment (n = 30), SCD control (n = 30), aMCI treatment (n = 30), aMCI control (n = 30), and a group of healthy adult subjects (n = 30) as a normal control (NC).
Results:
The primary outcome is the change in subjective and objective cognitive performance between baseline and postintervention visits (4 weeks after baseline). Secondary outcomes include changes in performance assessing from baseline, postintervention to follow-up (3 months after the intervention), as well as sleep, mood, and physiological parameters (including blood, urine, electrophysiology, and neuroimaging biomarkers).
Conclusion:
This study aims to provide evidence of the impact of light therapy on subjective and objective cognitive performance in middle-aged and older adults with SCD or aMCI. In addition, we will identify possible neurophysiological mechanisms of action underlying light therapy. Overall, this trial will contribute to the establishment of light therapy in the prevention of Alzheimer’s disease.
Keywords
INTRODUCTION
Background
Epidemiology of Alzheimer’s disease
The number of people with Alzheimer’s disease (AD) or other forms of dementia has been rapidly increasing as the number of elderly people grows [1]. The global prevalence of late-onset dementia is expected to triple [2], and the costs for dementia will be US $9.12 trillion in 2050 [3]. Numerous new drugs have failed in clinical trials [4–6], which stresses the need for safer and more alternative therapies for the disease, and the development of effective treatments for AD and dementia will have a very large socioeconomic impact.
Subjective cognitive decline and mild cognitive impairment are target populations for early intervention in AD
As pathophysiological changes may occur ten years or more before the primary clinical symptoms [7, 8], AD is regarded as a continuum that is divided into three stages: preclinical, mild cognitive impairment (MCI), and dementia [9]. Subjective cognitive decline (SCD) has been conceptualized to occur at the late stage of preclinical AD [10], where aberrant brain changes are present [11] in the absence of objective cognitive impairment. The manifestation of SCD is also acknowledged as one of the early signs of AD [12]. Along a numeric continuum, SCD and MCI are considered clinical stages 2 and 3, respectively [13]. Both SCD and MCI could increase the risk of objective cognitive decline and progression to dementia due to AD in the future [14, 15]. Moreover, persons with SCD or MCI (especially amnestic MCI, aMCI) harbor increased amyloid-β (Aβ) deposition [16, 17], neural dysfunction [18, 19], and gray matter volume reduction [20, 21] in brain regions typically affected in AD. Thus, these at-risk groups are recognized as suitable target populations for early intervention strategies [22, 23], aiming to protect against neuropathological alterations, recover functional and structural brain health, and maintain cognitive abilities as long as possible.
Light therapy in AD
Light not only guides humans’ and other mammals’ performance on cognitive tasks though vision but also exerts nonvisual effects that mediate task performance, as well as regulates circadian rhythms [24], melatonin production [25], changes in core body temperature [26, 27], sleep propensity [28], alertness [27, 29], and mood [30, 31]. Light therapy is the use of different strength levels and lengths of light to affect the suprachiasmatic nucleus and perihabenular nucleus by influencing intrinsically photosensitive retinal ganglion cells [32] with an aim to regulate learning and mood [33] and mediate circadian and sleep patterns [24]. Via a portable light source directly emitting to per eye, experimental results have shown that short-wavelength LED light between 400 and 525 nm had a safe and effective effect on human biorhythms [34, 35], while longer wavelengths (595 mm and 660 nm) were not effective in phase propulsion or in delaying circadian rhythm [34]. Hence, blue-green (500 nm) light may be as a suitable light therapy for cognitive-related disorders.
Light therapy as a nondrug therapy, which is a rapidly growing application for cognitive disorders [36], can promote connectivity between the thalamus and cortex and regulate circadian rhythms [29]. Light therapy is widely used for sleep disorders [37] and has also been used to explore effects on AD [38, 39]. Studies have shown that in patients with AD, light therapy can consolidate sleep at night [40], improve total wake time and sleep efficiency [41] and the quality of circadian activity rhythm [40, 42], attenuate cognitive deterioration and depressive symptoms [42], and reduce aggressive behavior [42, 43].
Given these observational findings, it has been hypothesized that light therapy in the early stages of AD may protect against cognitive function decline. The potential mechanism may be that light modulates cognition by influencing alertness-related subcortical structures (hypothalamus, brainstem, thalamus) and limbic areas (amygdala and hippocampus), which subsequently modulate activity in cortical areas [44]. Moreover, light is a central stimulator of circadian rhythms, sleep, and mood [24]. Therefore, rational use of light therapy may play a positive role in preventing and slowing the progression of AD.
Light therapy in SCD and MCI
Light therapy is thus proposed to open a new avenue in the protection and restoration of cognitive function. This expected benefit is of particular need in middle-aged and older individuals at risk for the development of dementia. It is, however, unknown whether the subjective and objective cognition-promoting effects of light therapy is detectable in persons with SCD or MCI and to what extent this effect may be attributed to the influence of light therapy on biomarkers of middle-aged and older adults.
Objective of this trial
We will conduct a randomized controlled trial with a 4-week light therapy in persons aged 45 years or older with aMCI (n = 60) and SCD (n = 60). A group of healthy adult subjects (n = 30) will be included as a normal control (NC). All groups are age-, sex-, and education-matched. The primary objective of this trial is to provide evidence of a beneficial impact of light therapy on subjective and objective cognitive performance (primary outcome) at the end of the intervention compared with the controls. Second, we aim to examine whether light therapy has positive effects on subjective and objective cognitive performance after an additional 3-month follow-up period without further therapy as well as on sleep and mood. These data will help to estimate the potential benefits of light therapy on “macroscopic” systems underlying cognition, sleep, and mood. Third, this trial will identify possible mechanisms of action underlying the proposed light therapy-associated benefits on subjective and objective cognition using blood- and urine-based biomarkers, electrophysiology, and neuroimaging parameters of brain structure and function. Finally, the study will assess potential moderators of the intervention effect, such as age-related neuropathology as well as genetic polymorphisms. These data will help to estimate the potential effects of light therapy on “microscopic” actions. Overall, this trial aims to foster the implementation of early intervention strategies in middle-aged and older individuals at risk of dementia due to AD.
METHODS: PARTICIPANTS, THERAPY, AND OUTCOMES
This will be a monocentric, randomized, controlled trial carried out at Chengdu Western Hospital, Chengdu University of Traditional Chinese Medicine (CDUTCM), University of Electronic Science and Technology of China (UESTC), Chengdu.
The trial will include 4 weeks of intervention with light therapy compared with the control condition (no treatment). The trial will compare outcomes from the five groups (i.e., NC, control of SCD, control of aMCI, treatment of SCD, and treatment of aMCI). Participants with SCD and aMCI will be randomized to the control or treatment group. Randomization will be performed blockwise with a 1:1 allocation ratio by age, sex, and diagnosis. The trial has been approved by the responsible institutional review board and will be carried out in compliance with institutional ethical standards and the Declaration of Helsinki.
Inclusion/exclusion criteria
The main inclusion criteria for potential participants in this study are as follows: 45–90 years of age. Right handedness and Mandarin-speaking. Presence of SCD according to the SCD diagnostic framework published by the international SCD-I working group in 2014 [10]: the expression of persistent subjective cognitive decline in the absence of objective cognitive impairment (i.e., does not meet the criteria for MCI [45] or dementia [46]), duration of at least 6 months, associated concerns (worries), affirmation to consults, no restrictions on activities of daily living, and not related to an acute event. Or presence of aMCI according to the criteria: memory complaints and meets any one of the following three criteria defined with the actuarial neuropsychological method proposed by Jak and Bondi [45]: 1) have impaired scores (defined as > 1 SD below the age-corrected normative means) on both measures in at least one cognitive domain (memory, language, or speed/executive function); 2) have one impaired score (defined as > 1 SD below the age-corrected normative means) in each of the three cognitive domains sampled (memory, language, or speed/executive function); and 3) have a score on the Functional Activities Questionnaire [47]≥9 indicating dependence in three or more daily activities. In this study, we set the long-term delayed recall and Auditory Verbal Learning Test-re-cognition tasks of the Auditory Verbal Learning Test-Huashan version (AVLT-H) [48] as the two measures in the memory domain, the Boston naming test [49] and the animal fluency test [50] as the two measures in the language domain, and part A and part B of the shape trails test [51] as the two measures in the executive function domain. Or meet the NC criteria: no self-reported persistent cognitive decline with the absence of objective cognitive impairment (i.e., does not meet the criteria for MCI [45] or dementia [46]). No hearing impairment or eye disease. Generally good condition and no other diseases that would affect the research. Not in a pregnant state, lactation period or child-bearing period (for example, women must be in menopause or have undergone a female sterilization operation at least 2 years before). Greater than 4 weeks of taking medicine that would have no impact on the research. Ability to provide written informed consent.
We will exclude potential participants meeting one or more of the following criteria: Dementia, according to the Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-V) [46]. Other neurological diseases that can cause cognitive decline (except for suspected early AD) including Parkinson’s disease, vascular dementia, Huntington’s disease, positive pressure hydrocephalus, brain tumors, progressive supranuclear palsy, epilepsy, subdural hema-toma, multiple sclerosis or brain trauma result-ing in neurological damage and other brain structural abnormalities. Other systemic diseases that can cause cognitive decline including Hashimoto’s encephalopathy, metabolic encephalopathy, paralytic dementia, anemia, hepatic encephalopathy, renal encephalopathy, etc. Clinical/baseline magnetic resonance imaging (MRI) demonstrates infection or other focal injury, multiple infarcts, or infarcts located in important memory brain areas. Pacemakers, aneurysm clips, artificial heart valves, ear implants, metal fragments, or foreign bodies in the eye, skin or body. A history of major depression, bipolar disorder, schizophrenia, and other psychiatric disorders (DSM-V diagnostic criteria) [46]. A history of alcohol or drug abuse/addiction in the past 2 years (DSM-V diagnostic criteria) [46]. The project cannot be completed due to any other systemic diseases or uncertain conditions.
All individuals included in the study must provide written informed consent and meet all eligibility criteria.
Therapy
Participants will receive 50 min continuous light therapy after they are awake in the morning over 4 weeks. Re-timer will be used as a light therapy device. Re-timer is a set of LED optical glasses that can emit soft green-blue light at a wavelength of 500 nm and will be provided by Flinders University and Samvardhana Motherson Group, Australia (http://re-timer.com/; Re-timer Pty Ltd, Adelaide, Australia).
Assessments of study measures
Measures assessed at baseline
The following participant characteristics will be collected or assessed at baseline: (a) demographic information, including age, sex, and education; (b) medical history; (c) information on family history focused on dementia and subtypes; and (d) behavioral measures of total cognitive function, mood and sleep. In addition, (e) physiological measures of blood Aβ, tau, and neurofilament light chain (NfL) will be measured using Quanterix’s single molecule array (Simoa™) technology [52], genotype information on apolipoprotein E (APOE) ɛ4 status will be measured using genotyping of blood-derived DNA [53], and brain structure and function will be assessed by electroencephalography (EEG) and multimodal MRI.
Outcome measures
Assessments of the primary study outcomes will be conducted at the postintervention visit (4 weeks after baseline). Secondary outcome measures will be assessed at the follow-up visit (3 months after the postintervention visit). Study outcomes will be collected in accordance with standardized operational procedures. Primary and secondary outcomes are summarized in Table 1.
Neuropsychological and subjective sleep tests
SCD-Q9, SCD-questionnaire 9; SCD-I, Subjective Cognitive Decline Interview; MMSE, Mini-Mental State Examination; MoCA-BC, Montreal Cognitive Assessment-Basic; CDR, Functional Activities Questionnaire; ADAS-cog, Alzheimer’s disease assessment scale-cognitive subscale; ADL, activities of daily living scale; FAQ, Functional Activities Questionnaire; AVLT-H, Auditory Verbal Learning Test; DST, digit span test; BNT, Boston naming test; AFT, animal fluency test; STT, shape trail test; SDMT, Symbol Digit Modalities Test; CDT, clock drawing test; HAMD, Hamilton Depression Scale; HAMA, Hamilton Anxiety Scale; NPI, Neuropsychiatric Inventory; MBI-C, Mild Behavioral Impairment Checklist; PSQI, Pittsburgh sleep quality index; RBDSQ, Rapid Eye Movement Sleep Behavior Disorder Screening Questionnaire; ESS, Epworth sleepiness scale; BQ, Berlin Questionnaire; HIS, Hachinski Ischemic Score; AD, Alzheimer’s disease; VaD, vascular dementia.
Primary endpoints. The primary endpoints of this trial are the changes in subjective and objective memory performance between the baseline visit (V1) and postintervention visit (V5). Subjective and objective memory performance was operationalized by the neurologist and will be assessed by the SCD questionnaire 9 and the Alzheimer’s Disease Assessment Scale cognitive subscale (ADAS-cog), respectively.
Moreover, the secondary outcomes, including the subjective and objective cognitive changes in performance from baseline to follow-up assessment (V6), behavioral markers (i.e., sleep, mood) and biomarkers (including blood, urine, electrophysiology and neuroimaging biomarkers), have been identified as sensitive outcome measures in older individuals at higher risk of AD [54–57].
Participant timeline
This trial will involve six phases for each participant: study enrollment, which will include a screening assessment (V0), a baseline visit (V1-V3), a 4-week light therapy period (V4), a postintervention visit (V5), and a follow-up visit (V6). Trial phases are described below and summarized in Fig. 1.
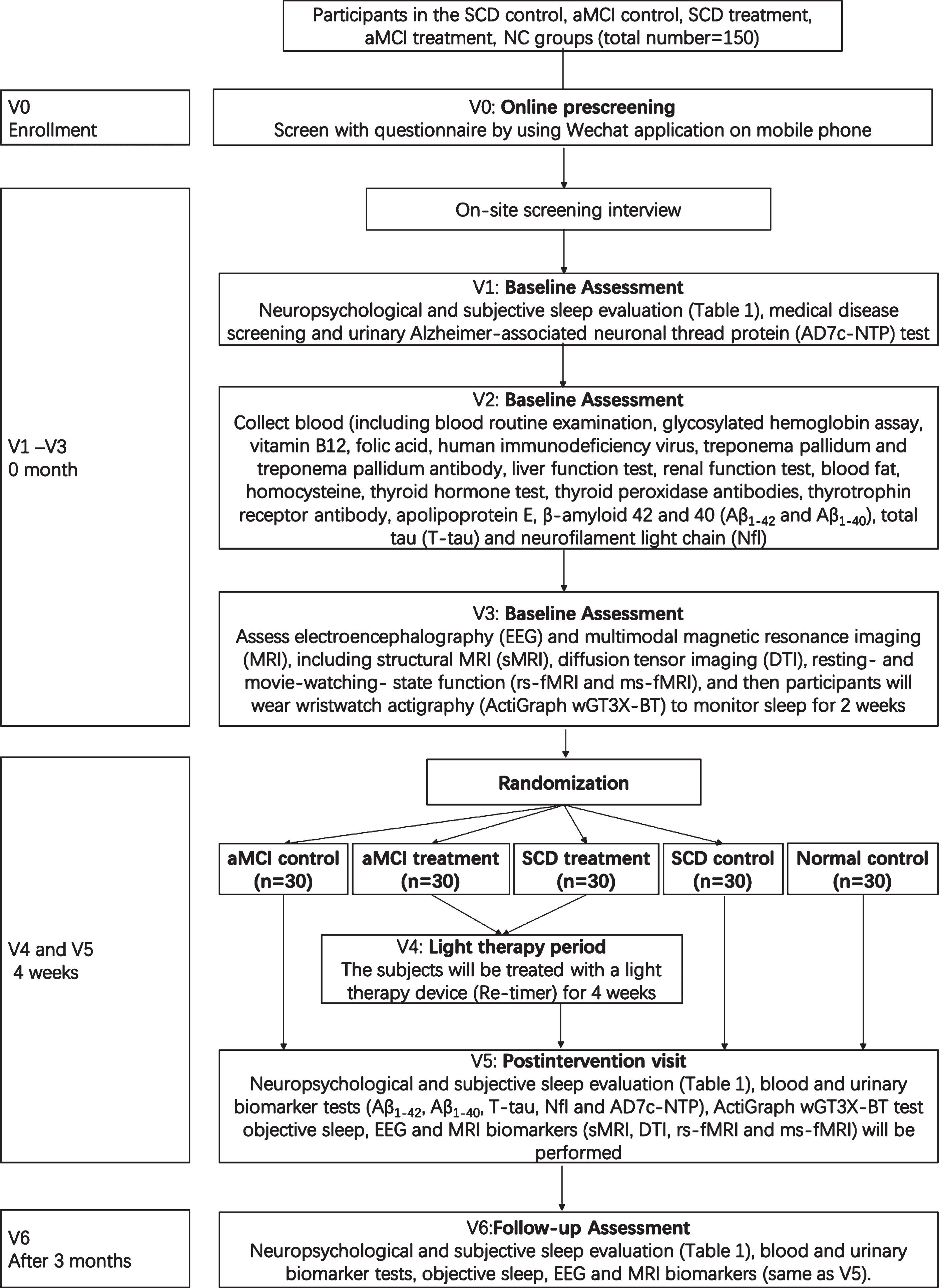
The overall research flow.
Enrollment (V0)
Individuals who are interested in participating in the study will undergo the following screening procedures to ensure study eligibility. A preliminary history of medical treatment, demographic data, MRI suitability and other information will be collected by using a WeChat mobile phone questionnaire. In this online questionnaire, the following information is provided:
(1) Name; (2) Date of birth; (3) Years of education (including night school, correspondence course, further study, etc.); (4) “Are you an ethnic minority?”; (5) “Are you left-handed (e.g., cooking, combing hair, writing, drawing)?”; (5) “Do you have some contraindication for magnetic resonance imaging (for example, pacemakers, metal fragments, etc.)?”; (6) “Have you ever had a stroke, anemia, gas poisoning, tumors, thyroid problems, and/or diabetes medical history before?”; (7) “Do you suffer from claustrophobia?”; (8) “Have you ever been told by your doctor that you have dementia?”.
Potential participants who meet the inclusion criteria through a prescreening assessment will be invited to the memory clinic of Chengdu Western Hospital for on-site screening interviews (V1).
Baseline visit (V1-V3)
Initial visit (V1): participants will visit the memory clinic of Chengdu Western Hospital for comprehensive neuropsychological evaluation, subjective sleep evaluation (Table 1) and medical screening, as well as urine collection for the Alzheimer-associated neuronal thread protein (AD7c-NTP) test. After meeting the inclusion criteria, participants will come for second visit (V2) to provide blood samples at CDUTCM. Routine blood examination, liver function, renal function, blood fat, blood glucose, glycosylated hemoglobin, vitamin B12, folic acid, homocysteine, thyroid hormone, thyroid peroxidase antibodies, thyrotrophin receptor antibody, human immunodeficiency virus (HIV) antibodies and Treponema pallidum-specific antibodies will be measured at CDUTCM. APOE assessments will be performed with a commercially available kit at Zhuhai Sinochips Bioscience Co., Ltd., Guangdong, China. Blood biomarker tests, that is, amyloid-β 42 and 40 (Aβ1–42 and Aβ1–40), total tau (T-tau), and NfL, will be performed with Simoa™ technology at G-Bio Biotech (Hangzhou) Co., LTD, Hangzhou, China. At the third visit (V3), subjects will visit the Qingshuihe campus of UESTC for the following assessments: EEG and multimodal MRI, including structural MRI (sMRI), diffusion tensor imaging (DTI) and resting-state and movie-watching-state functional (rs-fMRI and ms-fMRI). After MRI and EEG, participants will wear wristwatch actigraphy (ActiGraph wGT3X-BT) to monitor sleep for 2 weeks. The ActiGraph wGT3X-BT (https://actigraphcorp.com/actigraph-wgt3x-bt/) is a Wireless Bluetooth® Smart technology actigraph coupled with professional data analysis ActiLife desktop analysis software (version 6.13.4). It can effectively monitor and analyze sleep latency, number of awakenings, sleep time, sleep efficiency and other objective sleep indicators. The V2 and V3 visits could be interchangeable.
Light therapy period (V4)
The fourth phase (V4): after the last clinical test and examination, the subjects will be treated with a light therapy device (Re-timer) for 4 weeks. Each subject will wear the Re-timer for 50 min after they wake in the morning.
Re-timer (http://re-timer.com/) is a new wearable device in portable glasses and is intended to deliver light therapy for re-timing (or phase shifting) the human circadian rhythm. Re-timer emits highly efficient blue-green 500 nm light at an intensity of 506 lux (lm/m2) and 230μW/cm2, as measured at the surface of the eye (20 mm from the light source). The device administers light directly into the visual field at a fixed distance from the retina regardless of the head position and direction of gaze.
Postintervention visit (V5)
The postintervention visit (V5) will take place as quickly as possible after treatment ends. The as-sessments at the postintervention visit, including neuropsychological evaluation (Table 1), blood bio-marker (i.e., Aβ1–42, Aβ1–40, T-tau, and NfL) and urine biomarker (AD7c-NTP) tests, EEG and MRI biomarker (sMRI, DTI, rs-fMRI, and ms-fMRI) assessments, will be conducted over a maximum of 3 days and scheduled in close temporal proximity. In addition, participants will be asked to provide qualitative feedback on the intervention. The three control groups will be assessed in the same manner 4 weeks after the baseline visit.
Follow-up visit (V6)
The follow-up visit (V6) will take place 3 months after the light therapy treatment. This visit will include the following assessments: neuropsychological evaluation (Table 1) and blood, urine, EEG and MRI biomarker evaluation, which are the same as V5. The participants will again be asked to provide qualitative feedback on the intervention. Fig. 1 provides the overall research flow.
Sample size
In this study, the subjects with SCD or aMCI will be divided into two study arms (treatment or control, sample size 1:1) with four groups (i.e., control of SCD, control of aMCI, treatment of SCD, and treatment of aMCI) by a completely random method. At the same time, to obtain objective indicators from the normal group, the same tests will be conducted on the same number of normal people (i.e., NC), and the results will be taken as the standard value for the test scale. The outcome measure is the scores on the ADAS-cog.
The minimum difference between the clinical groups is estimated to be 1.30 points, and the difference between the treatment groups is estimated to be 1.2 points after treatment, with similar variance. For a bilateral test, with α= 0.05 and holding β=90%, the table shows that Zα= 1.96 and Zβ= 1.28. These values were substituted into the following formula [58]:
The calculation shows that the sample size n = 24.6402; that is, each group needs at least 25 people. Allowing for a maximum dropout rate of 10%, the number of people in each group needs to be set to n/10%, and 27.78 people can be calculated by substitution; that is, at least 28 people are needed in each group. Here, we set the sample size of each group as 30 people.
According to the foregoing and with the sample size of each group being 1:1, this meant that 120 patients with SCD or aMCI will be required to enroll in the experiment, including for treatment (30 SCD and 30 aMCI, respectively) and for control (30 SCD and 30 aMCI, respectively) groups, and 30 normal people are also needed to provide the reference data for the standard value; that is, 150 subjects are required in total.
Recruitment
Study participants will be recruited from memory clinics, neurologists, and the general population, as well as through advertisements in Chengdu, China.
METHODS: ASSIGNMENT OF THERAPY
Participants with SCD or aMCI will be randomly assigned to the light therapy or control groups. A blockwise (block size of 8) randomization sequence, stratified by age (45–59 and 60–90 years), dia-gnosis (SCD and MCI) and sex, will be generated using a computer-based algorithm (http://www.randomization.com/). Participant allocation will be performed at a 1:1 ratio by a study investigator without involvement in outcome assessments.
METHODS: MEDICAL ASSESSMENTS, NEUROPSYCHOLOGICAL ASSESSMENTS, SLEEP ASSESSMENTS, PHYSIOLOGICAL PARAMETERS COLLECTION
We will collect medical, neuropsychological, sleep, and physiological data from each participant. Details on the data collection procedures are provided below; the time points of collection are detailed in Table 1. To ensure the standardization of data collection, all study assessors will undergo systematic training. Most data will first be recorded on paper (see Table 1) and will be entered into electronic records after each study visit with ongoing quality checks throughout the study. Quality control will be ensured through regular data monitoring.
Medical assessments
The clinical assessments will include a structured medical history, physical examination, and routine laboratory tests (including routine blood tests, liver function, renal function, blood fat, blood glucose, glycosylated hemoglobin assay, vitamin B12, folic acid, homocysteine, thyroid hormone, thyroid peroxidase antibodies, thyrotrophin receptor antibody, HIV antibodies, and Treponema pallidum-specific antibodies).
Neuropsychological assessments
Neuropsychological tests will be conducted with subjects in a quiet, undisturbed environment. The neuropsychological battery will test subjective cognitive function, objective cognitive function (i.e., total cognitive function, memory, attention, language, executive functioning, and visuospatial functioning), mood, and neuropsychiatric and behavioral symptoms, as well as differentiate AD and vascular dementia. The tests include the SCD-questionnaire 9 [59], Subjective Cognitive Decline Interview [60], Mini-Mental State Examination [61], Montreal Cognitive Assessment-Basic [62], Clinical Dementia Rating [63], ADAS-Cog [64], activities of daily living scale [65], Functional Activities Questionnaire [47], AVLT-H [48], digit span test [66], Boston naming test [49], animal fluency test [50], shape trails test [51], Symbol Digit Modalities Test [67], clock drawing test [68], Hamilton Depression Scale [69], Hamilton Anxiety Scale [70], Neuropsychiatric Inventory [71], Mild Behavioral Impairment Checklist [72], and Hachinski Ischemic Score [73]. Neuropsychological tests will be administered at the baseline assessment (V1), postintervention visit (V5), and follow-up visit (V6) (see Table 1).
Sleep measurements
Self-reported sleep will be assessed using the Pittsburgh Sleep Quality Index (PSQI) [74] questionnaire, the REM Sleep Behavior Disorder Screening Questionnaire (RBDSQ) [75], the Epworth sleepiness scale (ESS) [76] and the Berlin Questionnaire (BQ) [77] at visits V1, V5, and V6 (Table 1). The PSQI questionnaire can assess subjective sleep quality, sleep latency, sleep duration, habitual sleep efficiency, sleep disturbances, sleep medication, and daytime dysfunction [74]. The RBDSQ provides a sensitivity of 0.96 and a specificity of 0.56 for screening rapid eye movement sleep behavior disorder [75]. The ESS provides a measurement of the subject’s general level of daytime sleepiness [76], and the BQ has been used to help identify patients at high risk of having sleep apnea [77].
Actigraphy (ActiGraph wGT3X-BT) will be used to quantify and record objective sleep. The device will be worn on the nondominant wrist for 2 consecutive weeks, starting after the EEG and MRI are assessed. Data will be evaluated with ActiLife software Version 6.13.4. ActiGraph and ActiLife provide information on the following parameters: sleep latency (SL), total sleep time (TST), sleep efficiency (SE), number and duration (minutes) of awakenings, and wake after sleep onset (WASO). For more detailed information, see the previous study [78].
Blood tests: APOE genotype, Aβ1–40, Aβ1–42, T-tau, and NfL
At least 400μL of whole blood will be collected from subjects by using a nonheparinized anticoagulant tube and then will be sent to the testing laboratory as soon as possible. If the samples cannot be sent immediately, they will be stored at 4°C and tested within 2 weeks. Repeated freezing and thawing will be avoided during the process. For genetic analyses, APOE characterization will be performed at Zhuhai Sinochips Bio-science Co., Ltd., Guangdong, China. The test will use polymerase chain reaction (PCR) with an APOE genotype test kit (gene chip) to amplify specific gene fragments, including the two APOE polymorphic sites (codons 112 and 158) [79]. The PCR parameters will be as follows: 50°C, 2 min; denaturation at 95°C for 15 min; 94°C, 30 s –65°C, 45 s for 45 cycles. Then, PCR products will be hybridized with the gene chip. Finally, a gene chip report system will be used to interpret the data [53].
Plasma will be collected in the morning (not fasting). EDTA plasma will be sampled, collected in the morning (fasting), aliquoted and frozen at –80°C according to standard procedures. For measurement of plasma Aβ1–40, Aβ1–42, total-tau, and NfL concentrations, one plasma aliquot will be transported on dry ice to G-Bio Biotech (Hangzhou) Co., Ltd., Hangzhou, China. All measurements will be performed using an in-house assay on the single molecule array platform (Simoa; Quanterix, Billerica, MA, USA), as previously described in detail [80].
All measurements will be performed by board-certified laboratory technicians who will be blinded to the clinical data.
Urinary AD7c-NTP test
Clean midstream urine specimens will be collected from all subjects in Eppendorf tubes. Levels of AD7c-NTP in urine samples will be measured using an enzyme-linked immunosorbent assay (ELISA) kit (Anqun Biological Technology Co. Ltd., Shenzhen, China) according to the manufacturer’s instructions. More detailed information is provided in the previous study [81]. AD7c-NTP is a potential biomarker that can be used for the diagnosis of AD and MCI [81, 82] and is related to the presymptomatic population with APOE ɛ4, which is a genetic risk of AD [83].
Magnetic resonance imaging
Brain image data collection
Magnetic resonance imaging data acquisition: The MRI examination will take place on a GE 3.0T MR750 scanner in the Qingshuihe Campus of UESTC. The MRI protocol will include structural and functional sequences. Each subject will undergo sMRI, DTI, and rs-fMRI scans. The specific scan parameters are presented in Table 2. In addition, a conventional MRI examination (T2 and FLAIR sequences) will be performed for each subject. The experienced neuroradiologist who will evaluate the MRI scans of each subject will look for gross anatomical abnormalities.
Neuroimaging data acquisition parameters
All MRI scans will undergo quality checks and will be evaluated quantitatively using state-of-the-art brain MRI software packages and toolboxes. During the scan, the subjects will be asked to close their eyes without thinking systematically, not to fall asleep, and to avoid movement of their head and body as much as possible.
Imaging data processing and analysis
sMRI data preprocessing and analysis. Analysis of gray matter volume and cortical thickness: For brain volume analysis, the images will be analyzed by Statistical Parametric Mapping Version 12 (SPM12; http://www.fil.ion.ucl.ac.uk/spm) and the Computational Anatomy Toolbox for SPM (CAT12; http://dbm.neuro.uni-jena.de/cat/). For cortical thickness analysis, images will be analyzed by FreeSurfer version 7 (https://surfer.nmr.mgh.harvard.edu/). The preprocessing steps will include bias correction, spatial standardization, tissue segmentation (gray matter, white matter and cerebrospinal fluid), extraction of inner and outer surfaces of gray matter, and definition and measurement of cortical thickness.
Structural brain network construction. The brain network and nodes will be defined to extract the morphological indices of each node, such as gray matter volume, cortical thickness and so on, and the structure of the brain network will be built. The specific morphological indices regarding any two nodes can be correlated among the subjects to obtain the corresponding morphological index correlation matrix after image binarization for the treatment groups and control groups after defining the brain structure of the network. The GRETNA source package (version 2.0.0, http://www.nitrc.org/projects/gretna/) will be used to calculate the topological properties of the whole-brain structural network, such as “small-world” features, global efficiency, node connectivity, etc. Comparisons of the differences in brain network topology between the treatment groups and the control groups will be assessed.
DTI data preprocessing and analysis
DTI data preprocessing. Raw DTI data will be conventionally processed using the Oxford Centre for Functional MRI of the Brain Software Library (FSL, version 6.0, http://www.fmrib.ox.ac.uk/fsl). It includes motion and eddy-current correction, brain extraction, and fractional anisotropy (FA) calculation.
Construction of a white matter network. Using the PANDA toolbox [84], fiber assignment by continuous tracking (FACT) for each participant’s whole brain fiber reconstruction will be conducted using the following parameter settings: start tracking seed points: FA > 0.2; terminate trace: FA < 0.2 or between two adjacent steps turning angle≥45°. For each subject, we will construct a structural network based on the fiber connections tracked throughout the brain. First, the brain will be divided into 90 brain regions in the original DTI space to define the nodes of the network. Second, the edges between nodes will be defined according to the results of whole-brain deterministic fiber tracking: when the fiber number between the two brain regions is≥3, it will be considered that there is a white matter connection between the two brain regions. The fiber number or average FA value between the two brain regions will be used to describe the connection strength between the two nodes. In this way, a binary network represented by a symmetric matrix with a size of 90×90 will be constructed for each subject.
fMRI data preprocessing and analysis
fMRI data preprocessing. The SPM12 source package and DPARSF software (version 5.1, http://www.restfmri.net/forum/DPARSF) will be used to routinely preprocess all rs-fMRI data. The main steps will include [85] removing the first 10 time points of each subject, slice timing and head motion correction, normalization, smoothing, removing the linear tread, and filtering. Then, regression analysis will be used to reduce the influence of subjects’ age, sex, education, head movement parameters and other brain tissue signals (including mean whole-brain signals, white matter signals and CSF signals).
Construction of the functional brain network. The cortical regions of the whole brain will be seg-mented with the automated anatomical labeling (AAL) template to obtain the division of brain regions. The average time series of each region will be extracted from the preprocessed data. For each subject, a functional connection matrix will be established by calculating the Pearson correlation coefficients of the average time series across brain regions. After binarization of the matrix, the resting brain functional binary network of each subject will be obtained. The correlation coefficient will be used to assign values to the edges of the network to obtain the functional weighted network. Then, through the GRETNA software package, the topological properties of the brain functional network will be calculated, including global properties (such as cluster coefficient, characteristic path length, “small world” properties, etc.) and local properties (node connectivity, medium centrality, node efficiency, etc.). In addition, the measures of local connectivity, including the four-dimensional (spatiotemporal) consistency of local neuron activity (FOCA) [86] and local functional connectivity density (local FCD) [87], will be calculated using SPM12 as implemented in the Neuroscience Information Toolbox (http://www.neuro.uestc.edu.cn/NIT.html, NIT) software package [88]. The nonlinear relations between multidimensional clinical and fMRI indices will be calculated using eigenspace maximal information canonical correlation analysis (emiCCA) [89].
EEG data collection and analysis
The EEG data will be collected on an EEG16-BT and/or EEG32-BT EEG amplifier (BORUIEN, China). The placement of the 16 electrodes will be as follows: Fz, Fp2, F3, F4, C3, C4, Cz, Pz, Fc4, Fc3, F7, F8, T3, T4, T5, and T6 [90]. The 32 electrodes will be located according to the 10–20 system, and more detailed information is provided in a previous study [91]. The impedance for all electrodes will be kept below 5 KΩ. The EEG signals will be digitized with a sampling rate of 1000 Hz.
EEG will be preprocessed using a stable EEG preprocessing tool integrated by a cloud WeBrain platform (http://webrain.uestc.edu.cn/). The preprocessing will include the quality assessment of raw EEG data, passband (1–40 Hz), artifact removal using electro-oculogram regression and residual artifact removal method (ICA-based multiple artifact rejection algorithm) [92], bad channel interpolation realized by the reference electrode standardization technique (REST) [93, 94], quality assessment of preprocessed data and marking residual bad blocks. Then, clean EEG data with the REST reference will finally be obtained. Finally, various EEG indices, including the power spectrum, EEG network and topology, will be calculated using EEG tools on the WeBrain platform. More details about these tools can be seen in the tool instructions, which can be downloaded from the WeBrain website (http://webrain.uestc.edu.cn/documentation.html).
Data management and monitoring
All data will be stored on a secure server with two back-up copies on external hard drives. To certify excellent data quality, good clinical practice (GCP) monitoring will be held on a regular basis. Paper-based form documents will be digitized, and the original copies will be stored in locked filing cabinets in the archive of GCP and managed by a department staff member who is external to the research team. All randomized participants will be referenced on all forms by participant ID. A password-protected spreadsheet stored on the secure server will link all participant names to ID codes so that reidentification can occur if required.
Statistical analysis
The designated statistician will conduct statistical analyses of the primary outcome. The study investigators will carry out statistical analyses of the secondary outcomes and additional exploratory analyses.
The primary outcome will be analyzed by using an “intention to treat” (ITT) method, which includes all participants randomized into the trial, regardless of the length of intervention. Then, a “per protocol” analysis will be carried out, including only those participants who finished the 4-week intervention period. We will compare the differences in the changes in subjective and objective cognitive performance between baseline (V1) and the end of intervention (V5). An ANCOVA model with the change in outcomes (V5-V1) as the dependent variable will be used, while the intervention group will be used as the independent variable, and baseline cognitive performance as well as sex, age and education will be used as covariates. The secondary outcomes will be analyzed by using comparable statistical methods. For example, an analysis of changes in subjective cognitive performance between baseline (V1) and follow-up assessment (V6) will be carried out by means of ANCOVA. In this model, the change in subjective cognitive performance (V6–V1) will represent the dependent variable, with the intervention group as the independent variable and baseline scores, sex, age and education inserted as covariates. Regarding outcomes with MRI and EEG indices, the ANCOVA model will perhaps also contain specific covariates such as head motion and brain volume.
In the case of missing primary endpoints, multiple imputation methods will be performed in the primary analysis. By integrating demographic or biological factors (i.e., APOE ɛ4 carriers) into statistical models as covariates (or as interactive terms with the intervention group), we will further investigate whether these variables are associated with primary and secondary endpoints and/or modulate the response to the light intervention. We will test hypotheses in a two-sided test at alpha = 0.05 using the ITT data set with multiple imputation.
DISCUSSION
This is a monocentric, randomized, and controlled trial to determine the potential beneficial impacts of 4 weeks of continuously wearing blue-green (500 nm) LED glasses in the morning for light treatment 50 min per day on subjective and objective cognitive performance in mid-age or older individuals at risk for the development of AD. We will mainly assess cognitive performance between baseline and postintervention visits (4 weeks after baseline) and follow-up assessments (3 months after the intervention), as well as changes in outcome measures of sleep, mood and physiological parameters (including blood, electrophysiology, and neuroimaging biomarkers), to evaluate further benefits of the intervention and to further identify possible neurophysiological mechanisms of action underlying light therapy.
The target groups of our trial comprise cognitively unimpaired mid-aged and older individuals with SCD and aMCI. Given that these populations are at higher risk of objective cognitive decline and clinical progression [95, 96], it is the key goal of this study to contribute to the development of effective prevention strategies. The daily solar cycle allows organisms of mammals to synchronize their circadian rhythms and sleep–wake cycles to the correct temporal niche [97]. The central circadian pacemaker is found in the suprachiasmatic nucleus [98]. Animal experiments showed that light mediates hippocampal learning and mood by projecting intrinsically photosensitive retinal ganglion cells to the suprachiasmatic nucleus and perihabenular nucleus, respectively [33]. In addition, nighttime-light could induce depression by mediating the intrinsically photosensitive retinal ganglion cells⟶dorsal perihabenular nucleus⟶nucleus accumbens pathway [31], while daylight is considered a “drug” to treat many clinical conditions, such as winter and other depressions and circadian sleep disorders [99]. By measured the normal human saliva, previous studies found that maximum suppression of melatonin following 1 h of light was 44%with intensity of 500 lux [100] and the minimum light intensity decreased as duration of exposure increased [101]. This current trial uses Re-timer, which emits highly efficient blue-green 500 nm light at an intensity of 506 lux (lm/m2) and 230μW/cm2, as light source and wears it for 50 min, which will suffice to suppress the melatonin secretion follows to the previous studies [100, 101]. Based on existing findings in animals and in humans [27, 40–43], morning short-wavelength light therapy may be expected to protect cognitive performance and benefit sleep and mood disorders, but at present, there are few experimental studies on the effect of blue-green (500 nm) light in light therapy on preclinical and prodromal AD patients, such as SCD and aMCI patients, regarding cognition, sleep, and mood disorders.
The strengths of the trial are as follows: 1) One advantage of this study is focusing on a variety of biomarkers (e.g., clinical, blood, urine, EEG, and imaging markers) to observe the changes in SCD and aMCI patients to investigate the effects of light therapy for improvement. Study outcomes will provide data supporting the clinical application of light therapy on biomarkers of AD and improvement. 2) Studies have shown that the effects of blue-green LED light on the body’s biorhythms are safe and effective. In addition, there are few domestic clinical studies on the regulation of human biorhythms on blue-green light therapy, and this study can offer supporting evidence. 3) The wearable device (Re-timer) is a portable family device with a reasonable price, and this trial will provide research support for the sleep device regarding use in the daily life of the family.
The limitations of this trial include the time period of the intervention, which is relatively short given the slow evolution of AD pathogenic processes in nondemented older individuals [22], and the monocentric trial is inadequate regarding the number of participants compared to a larger multicenter study. In addition, our participants will mainly be from Chengdu, Sichuan Province, which could not represent SCD and aMCI patients nationwide. Last, it is important to highlight that a multicenter trial implementation is not financially feasible. Pending the positive results of this trial, public funding for a large and multicenter trial will be applied for, to move this intervention into routine clinical practice.
Overall, our trial aims to contribute to the establishment of an effective and well-tolerated wearable and household type of light therapy to promote cognitive and brain health in older individuals at higher risk of dementia. A positive outcome with regard to behavior performance, as well as physiological parameters, in the light therapy group may initiate a large multicenter trial with a profound impact on public health, patients with SCD or aMCI and their families.
Trial status
Recruitment of participants started in September 2019. Because of the COVID-19 pandemic, we hope recruitment can be finished in December 2021, and the last follow-up is scheduled for May 2022.
Footnotes
ACKNOWLEDGMENTS
This work is supported by the National Natural Science Foundation of China (Nos. 81861128001), the ‘111’ project (B12027), the Sichuan Science and Technology Program (Nos. 2018JZ0073, 2019YJ0179 and 2018SZ0192), the CAMS Innovation Fund for Medical Sciences (2019-I2M-5-039), National Key R&D Program of China (2020YFC2003100, 2020YFC2003104), the Chengdu Municipal Health and Family Planning Commission Research Project (2018067, 2020021) and the “Xing-lin Scholars” Project of Chengdu University of Traditional Chinese Medicine (grant no. QNXZ2018004).