
Abstract
Dementias, including the type associated with Alzheimer’s disease (AD), are on the rise worldwide. Similarly, type 2 diabetes mellitus (T2DM) is one of the most prevalent chronic diseases globally. Although mechanisms and treatments are well-established for T2DM, there remains much to be discovered. Recent research efforts have further investigated factors involved in the etiology of AD. Previously perceived to be unrelated diseases, commonalities between T2DM and AD have more recently been observed. As a result, AD has been labeled as “type 3 diabetes”. In this review, we detail the shared processes that contribute to these two diseases. Insulin resistance, the main component of the pathogenesis of T2DM, is also present in AD, causing impaired brain glucose metabolism, neurodegeneration, and cognitive impairment. Dysregulation of insulin receptors and components of the insulin signaling pathway, including protein kinase B, glycogen synthase kinase 3β, and mammalian target of rapamycin are reported in both diseases. T2DM and AD also show evidence of inflammation, oxidative stress, mitochondrial dysfunction, advanced glycation end products, and amyloid deposition. The impact that changes in neurovascular structure and genetics have on the development of these conditions is also being examined. With the discovery of factors contributing to AD, innovative treatment approaches are being explored. Investigators are evaluating the efficacy of various T2DM medications for possible use in AD, including but not limited to glucagon-like peptide-1 receptor agonists and peroxisome proliferator-activated receptor-gamma agonists. Furthermore, there are 136 active trials involving 121 therapeutic agents targeting novel AD biomarkers. With these efforts, we are one step closer to alleviating the ravaging impact of AD on our communities.
Keywords
INTRODUCTION
Dementias, including the type associated with Alz-heimer’s disease (AD), are on the rise worldwide. It is estimated that about 50 million people worldwide have dementia, with AD being the most common form. In the United States alone, there were 5.8 million cases of AD in 2020 [1, 2]. Sporadic AD is a progressive neurologic disease that usually affects individuals in later life. Beginning as mild memory loss, AD progresses with continued cognitive, behavioral, and social decline. Hallmarks of AD include amyloid plaques, composed of amyloid-β (Aβ) aggregates, and tau protein organized into neurofibrillary tangles (NFTs). Both protein structures have neurotoxic effects and disrupt cell communication. Over time, atrophy of the brain, neuron death, and severe memory impairment occur [1].
Similarly, diabetes is one of the most prevalent chronic diseases globally, affecting more than 422 million people worldwide and 34 million Americans [3, 4]. T2DM accounts for the majority of these cases [4]. T2DM involves a decreased response to insulin, a phenomenon known as insulin resistance. In response, the pancreas works to create more insulin. However, over time, insulin production does not meet the demand to keep blood glucose under control, resulting in insulin deficiency and elevated glucose levels. Prolonged hyperglycemia, in turn, contributes to other serious diseases, such as vision loss, heart disease, and kidney disease [3].
Previously perceived to be unrelated diseases, co-mmonalities between T2DM and AD have more recently been discovered. Several studies have evaluated the association between AD and T2DM with mixed results. In T2DM, brain atrophy, changes in brain volume, and alterations in white matter micro-structure and connectivity are seen. Studies show that more than 40%of T2DM patients have intermediate to severe AD pathology in their brains at the time of death [5]. However, other examinations of autopsied brain samples have found no relationship between T2DM and AD pathology [6]. Studies show that T2DM increases the risk of developing AD by 1.3–2.3-fold [7]. Meta-analyses by Anstey et al. (2019) and Rochoy et al. (2019) found that diabetes is a risk factor for AD, with an estimated relative risk (RR) of 1.39 (95%confidence interval [CI], 1.16–1.66) and 1.53 (95%CI, 1.42–1.63), respectively [8, 9]. Another analysis of 24 studies found that diabetes increased the risk of AD (RR, 1.43; 95%CI, 1.25–1.62) [10]. In a systematic review and meta-analysis by Ninomiya et al. (2019), the pooled hazard ratio (HR) for AD in patients with T2DM was 1.6 (95%CI, 1.4–1.8) [11]. On the other hand, two studies showed a negative association between T2DM and AD (odds ratio [OR], 0.84; 95%CI, 0.78–0.90) [12].
Although mechanisms and treatments are well-est-ablished for T2DM, there remains much to be discovered regarding AD. Recent research efforts have further investigated factors involved in the etiology of AD. In this review, published articles from 2018 to 2021 are examined to highlight shared mechanisms between these two disease states. They include insulin signaling dysfunction and resistance, inflammation, oxidative stress, mitochondrial dysfunction, advanced glycation end products (AGEs), and amyloid deposition. The roles of gut and microbiota, impaired vasculature and the blood-brain barrier (BBB), genetics, and vitamin and mineral levels are also discussed. The use of antidiabetic medications in AD and research into other therapies are also addressed.
MECHANISMS
Insulin signaling and resistance
The process of insulin signaling (Fig. 1) involves insulin secretion from pancreatic β-cells and binding of insulin to the insulin receptor, which causes phosphorylation of insulin receptor substrates 1 and 2 (IRS-1 and IRS-2). Phosphorylated IRS activates and phosphorylates phosphoinositide 3-kinase (PI3K), which then phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) to generate phosphatidylinositol (3,4,5)-triphosphate (PIP3) [13, 14]. PIP3 activates phosphoinositide-dependent protein kinase 1 (PDK-1), which phosphorylates and activates protein kinase B (AKT) [14]. AKT controls translocation of glucose transporter 4 (GLUT4), the main glucose transport protein, to the plasma surface. AKT also regulates activity of glycogen synthase kinase 3β (GSK3β) and mammalian target of rapamycin (mTOR) [13, 14]. Disturbances in this insulin signaling cascade have been identified in both AD and T2DM.
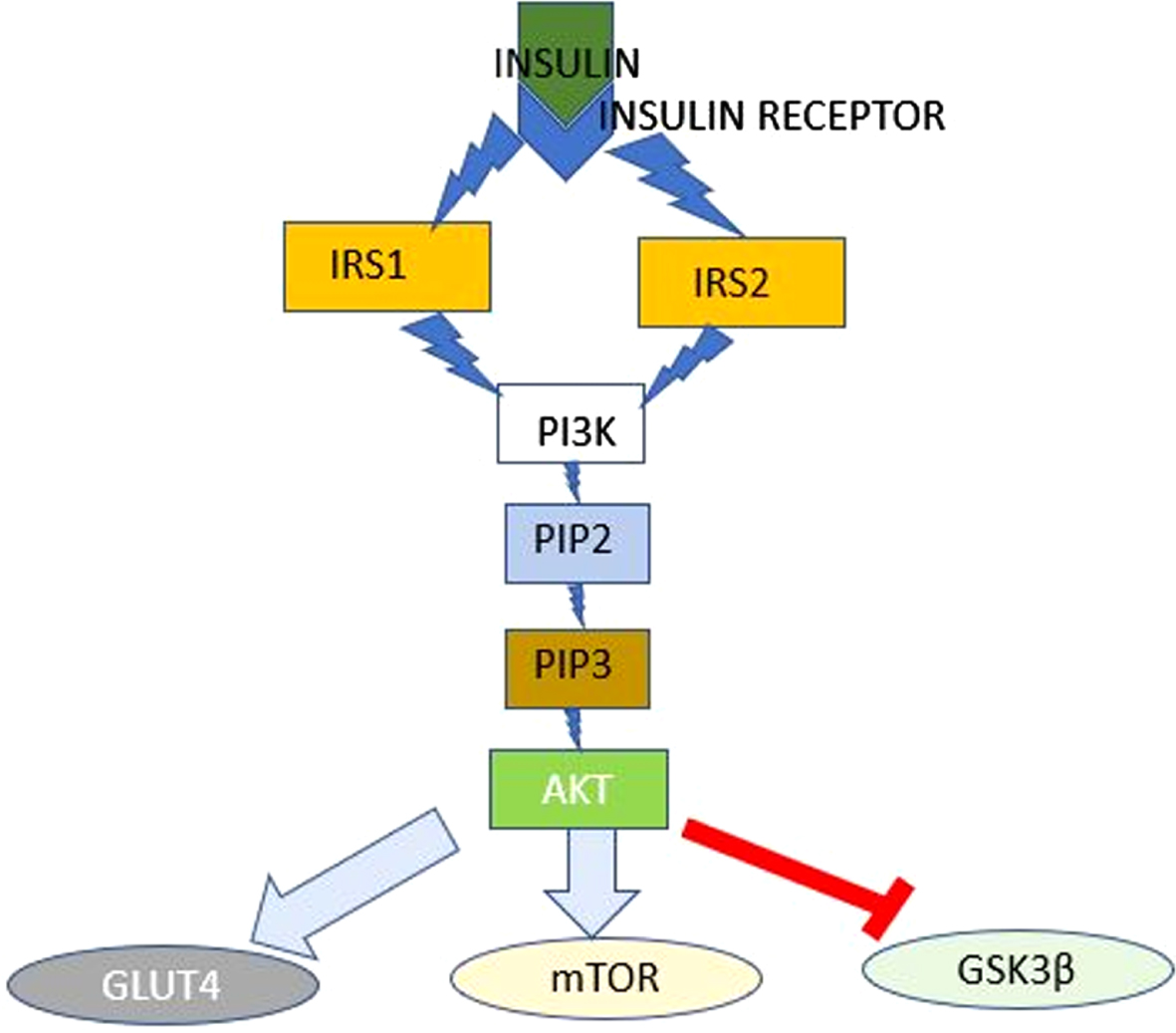
Insulin Signaling Pathway. The Insulin Signaling Pathway: Insulin binding at its receptor initiates a cascade of events that culminate in the regulation of the activities of GLUT4, mTOR and GSK3β. AKT, protein kinase B; GLUT4, glucose transporter 4; GSK3β, glycogen synthase kinase 3β; IRS1, insulin receptor substrate 1; IRS2, insulin receptor substrate 2; mTOR, mammalian target of rapamycin; PDK-1, phosphoinositide-dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol (4,5)-bisphosphate (PIP2); PIP3, phosphatidylinositol (3,4,5)-triphosphate.
Insulin resistance is the decreased ability of insulin to cause glucose uptake from the blood, leading to increased insulin secretion from the pancreatic β-cells, hyperinsulinemia, and hyperglycemia [15]. As the pancreatic β-cells lose function, not enough insulin is produced to maintain normal blood glucose [16]. Insulin resistance is a major contributor to the elevated blood glucose levels of T2DM and is observed in patients with AD, perpetuating the idea of AD being a “type 3 diabetes mellitus” [17]. Insulin also plays a role in glucose metabolism in the brain, which contributes to the processes of synaptic plasticity, transmission, neurogenesis, and secretion of neurotransmitters, which in turn, influence cognitive function [18]. Since insulin is also believed to have a neuroprotective effect, insulin resistance may potentiate neurodegeneration and cognitive impairment seen in both diseases [14, 17].
The hippocampus, hypothalamus, olfactory bulb, cerebellum, amygdala, and cerebral cortex have a high concentration of insulin receptors [14, 17]. Impaired insulin signaling caused by insulin resistance prevents binding of insulin to its receptor and the downstream processes leading to neural development [17]. Additionally, insulin resistance or high levels of circulating insulin downregulates insulin receptors at the BBB. This leads to decreased ins-ulin permeability, decreased insulin in the central nervous system (CNS), and potentially reduced insulin-induced neural and glial activity [6, 19]. An association between chronic diabetes and neurodegeneration has been observed, with about 29%of T2DM patients displaying severe cognitive decline [17]. Furthermore, studies have shown that reduced insulin response was associated with an increased risk of developing AD [20]. Neuronal insulin resistance may also contribute to AD as downregulation of insulin receptors was found in the brains of AD patients [21]. Altered peripheral glucose regulation characterized by hypoinsulinemia, impaired insulin secretion, or hyperinsulinemia and insulin resistance is observed in many AD patients [20]. Studies have shown reduced expression and activation of insulin receptor and insulin growth factor 1 receptor (IGF-1R) in the brains of AD patients [14]. Reverse transcription polymerase chain reaction and western blotting of brain tissue samples of AD patients showed a ∼15-fold decrease in insulin receptor mRNA in the hippocampus and a ∼50%decrease in insulin receptor protein levels [22]. IRS-1 and IRS-2 were also significantly downregulated in the brains of AD patients [18]. Peripheral insulin resistance may also lead to insulin resistance in the brain and reduction in glucose uptake. Insulin increases translocation of glucose transporters to the plasma membrane, regulating glucose availability [20]. In early AD, glucose utilization by the brain decreases [21]. In postmortem human AD tissue and some AD mouse models, decreased glucose transporter 1 (GLUT1) and the neuronal glucose transporter, GLUT3, have been observed [20]. Multiple studies have also shown that triple transgenic AD (3xTg-AD) mice have impaired glucose tolerance [20].
Insulin resistance also promotes Aβ accumulation as demonstrated by the presence of elevated brain Aβ with induction of insulin resistance in transgenic AD mice and diabetic obese mice given a high fat diet [14]. Insulin and Aβ are both substrates of insulin degrading enzyme (IDE). Hyperinsulinemia, which can be seen in both diseases, causes IDE to become saturated, preventing degradation and clearance of Aβ. This results in Aβ accumulation, formation of NFTs, and neurodegeneration [23]. Aβ oligomers (AβOs) also perpetuate disrupted insulin signaling by reducing insulin receptor levels and activity [14]. AβOs cause insulin receptor movement from the cell surface into the cytoplasm, preventing interaction with insulin, IRS, and insulin growth factor (IGF) needed to initiate insulin signaling [22, 24]. By disrupting insulin signaling, Aβ promotes a positive feedback loop that increases amyloid-β protein precursor (AβPP) processing and further Aβ formation [17]. AβOs also induce tumor necrosis factor alpha (TNF-α) secretion by microglia and astrocytes, which contributes to this decreased neuronal insulin signaling by stimulating c-Jun N-terminal kinase (JNK) [22]. JNK in turn phosphorylates IRS-1, preventing the activation of PI3K and AKT needed for further insulin action [22]. In the brains of patients with AD, AD animal models, and cognitive impairment-induced T2DM models, elevated IRS-1 phosphorylation was observed [25]. Normally, AKT phosphorylates GSK3β, a protein serine/threonine kinase that phosphorylates the enzyme glycogen synthase and has a role in apoptosis and long-term potentiation required for synaptic maintenance and plasticity. This phosphorylation inactivates GSK3β and prevents GSK3β-induced tau hyperphosphorylation [24, 26]. However, decreased insulin signaling causes reduced AKT activation and in turn, GSK3β overactivation, leading to hyperphosphorylation of microtubule-associated protein tau (Fig. 2) [24, 26]. This concept was supported by a study showing that AD brains had reduced expression of activated AKT, insulin receptor mRNA, and IRS-associated PI3K [27].
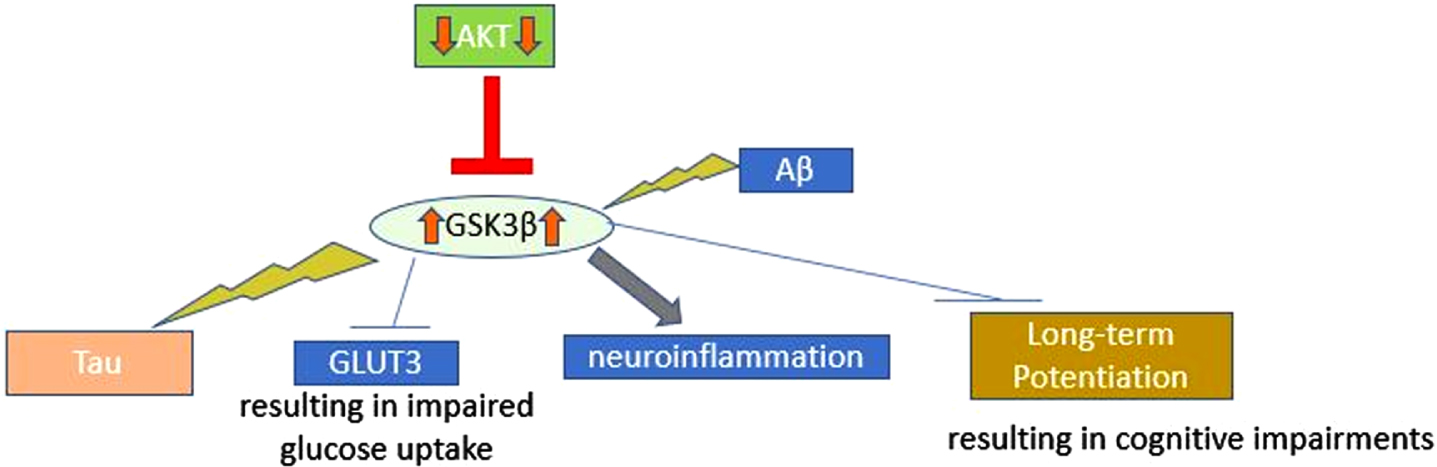
GSK3β Overactivation. GSK3β Overactivation: Activation of GSK3β leads to phosphorylation of tau, inhibition of GLUT3, inhibition of long-term potentiation (resulting in cognitive impairments), and promotion of neuroinflammation. Aβ, amyloid-beta; AKT, protein kinase B; GLUT3, glucose transporter 3; GSK3β, glycogen synthase kinase 3β.
GSK3β dysfunction has also been associated with the development of T2DM and AD. The result is microtubule instability as hyperphosphorylated tau disassociates from the microtubule assembly and leads to paired helical filamentous tau, which is a major component of the NFTs that are characteristic of AD [24, 26]. In postmortem AD brains, this increased GSK3β activity has been observed [18]. Activation of GSK3β is further promoted by Aβ and causes neuroinflammation and reduction in acetylcholine synthesis [18]. GSK3β also inhibits GLUT3, preventing glucose uptake into the cells [17]. GSK3β overexpression can impair cognition and long-term potentiation by decreasing postsynaptic density and volume in the hippocampus [26]. In transgenic mice, GSK3β overexpression impaired spatial recognition and memory [28].
mTOR is a protein kinase involved in the insulin pathway, energy metabolism, and the regulation of cell proliferation, transcription, and translation [29, 30]. Autophagy is a cellular process used to remove abnormally aggregated and misfolded proteins and damaged organelles. It is believed that dysfunction in autophagy caused by mTOR signaling leads to tau hyperphosphorylation [29]. In T2DM, mTOR activation is enhanced and leads to tau hyperphosphorylation [29]. Aβ also hyperactivates mTOR as shown by the overactivation of the PI3K-AKT-mTOR pathway seen in postmortem examination of AD brains [29, 31]. Further supporting the claim that mTOR is involved in tau pathology is that rapamycin, an mTOR inhibitor, has demonstrated reduction of Aβ and tau in 3xTg-AD mice [29]. Late-phase long-term potentiation is also reduced with mTOR hyperactivation [32].
Inflammation, oxidative stress, and mitochondrial dysfunction
Overproduction of pro-inflammatory cytokines, such as TNF-α, interleukin-1β (IL-1β), IL-6, IL-18, and leptin from the adipose tissues of diabetic patients can impair insulin signaling and increase the risk of insulin resistance [14]. Hyperinsulinemia also promotes increased cerebral levels of pro-inflammatory cytokines [14]. Recruited macrophages further enhance the secretion of IL-1β, IL-6, and IL-10 [33]. Similarly, in AD, IL-6, TNF-α, and C-reactive protein prevent the movement of Aβ out of the CNS, causing accumulation and microglial activation [34]. CSF and brain tissue of AD patients and AD mouse models show increased cytokine levels, microglia, and astrocytes [20, 35]. Microglia further increase pro-inflammatory cytokines, such as TNF-α, IL-6, IL-1β, interferon-γ, and macrophages [24]. Aβ production and deposition can be affected by IL-1, which regulates AβPP expression, synthesis, and processing [35]. Oxidative stress increases the activation of β- and γ-secretases responsible for production of Aβ from AβPP [35]. Neuroinflammation also causes cholinergic dysfunction, which leads to neuronal injury [24].
Mitochondria are essential for cell function, energy generation, apoptosis, redox signaling, reactive oxygen species (ROS) and reactive nitrogen species production, steroid and neurotransmitter synthesis, innate immunity, and autophagy [36, 37]. Mitochondrial dysfunction is a common feature of T2DM and AD [37]. In T2DM, ATP production and mitochondrial function in pancreatic β cells are decreased, which can alter insulin secretion [38]. Hyperglycemia in T2DM enhances ROS, which mediates lipid peroxidation and causes alterations in plasma membrane integrity and β-cell dysfunction [39]. Cell injury from fission-mediated fragmentation of mitochondria has also been seen in hyperglycemic conditions [37]. Similarly, Aβ overproduction in AD increases mitochondrial fragmentation and oxidative injury to the membrane, disrupting lipid polarity and protein mobility and inhibiting enzymes of mitochondrial respiration. This increases permeability of the mitochondrial membrane and ATP depletion [35]. Studies have found a reduction in oxygen consumption and altered mitochondrial structure, size, and number in AD [37, 40]. In both T2D-like and 3xTg-AD mice, brain mitochondrial abnormalities, such as defects in turnover, biogenesis, and function, as well as redox imbalance, have been seen [36].
Oxidative stress has been observed in hyperglyce-mia, leading to pathways that upregulate inflammatory cytokines. These circulating cytokines facilitate apoptosis, decrease neurogenesis, and reduce synaptic function [34]. In AD, oxidative stress is involved in tau hyperphosphorylation and Aβ accumulation [41]. Misfolded Aβ and tau, components of AD, destabilize the mitochondrial membrane and disrupt enzymes in mitochondrial metabolism [38]. Microtubule-associated protein, including tau, enters the synapse via axons and helps to transport mitochondria. Tau hyperphosphorylation alters this transport of mitochondria, causing energy deficiency in the synapse. AD mouse models have shown that hyperphosphorylated tau and mitochondrial dysfunction are linked to synapse loss and reduced cognition [38].
Studies have shown that hyperglycemia and oxidative stress can cause accumulation of mitochondrial DNA mutations, which leads to mitophagy-resistant mitochondria [37]. Mitophagy is the removal of damaged mitochondria via mitochondrial biogenesis and autophagy. Alterations in mitophagy have been shown to contribute to impaired insulin synthesis, secretion, and sensitivity. Similarly, dysfunction in mitophagy and accumulation of damaged mitochondria has been observed in AD, leading to neurodegeneration, synaptic damage, and neuronal loss [37]. Events leading to mitochondrial dysfunction are summarized in Fig. 3.
Advanced glycation end products
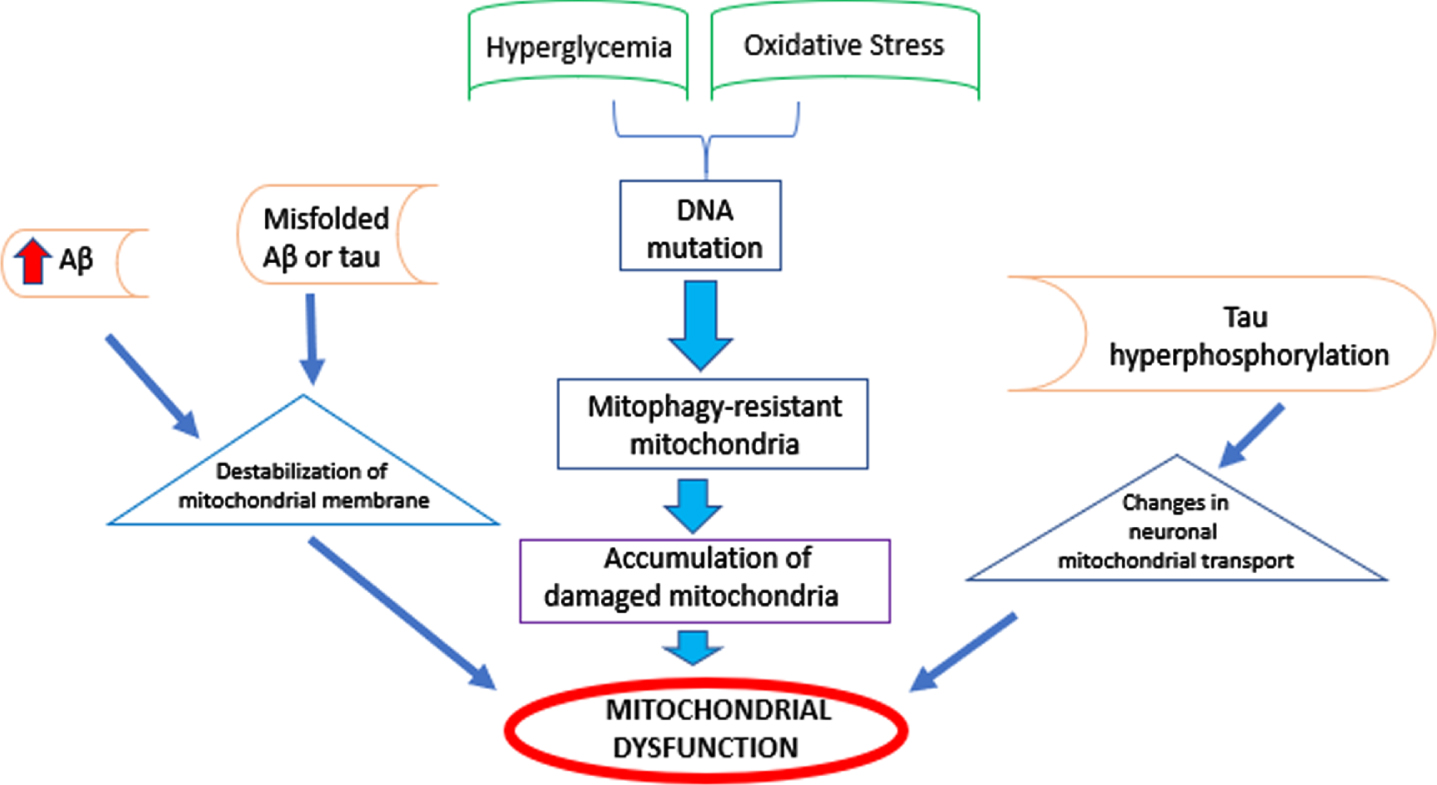
Mitochondrial Dysfunction. Factors contributing to alterations in number, structure, or size of mitochondria in T2DM or AD. These events culminate in mitochondrial dysfunction.
AGEs are products of reactions between proteins or lipids with sugars that accumulate with older age in both T2DM and AD patients [17, 27]. When AGE interacts with AGE-receptor (RAGE), inflammatory mediators and the nuclear factor-kappa B (NF-kB) pathway are activated [39]. AGEs are believed to increase ROS, causing synaptic dysfunction, damage to nerve terminals, and destabilization of lipids, proteins, DNA, and RNA [24]. The resulting oxidative stress results in intracellular and tissue damage and Aβ production [20, 42]. AGEs can also cause downregulation of the sirtuin 1 (SIRT1) protein, causing improper binding of insulin receptor to phosphorylated IRS-1 and ineffective insulin signaling [17]. Thus, in vivo studies of mice fed AGE showed cognitive dysfunction and pathology resembling metabolic syndrome [24, 27].
RAGE was suggested to be a receptor for Aβ and a mediator of Aβ-induced endoplasmic reticulum stress [27]. The role Aβ plays in BBB integrity, regulation of tight junctions, and apolipoprotein E (APOE)-induced BBB abnormalities is believed to be mediated by RAGE [27]. Therefore, the interaction of Aβ with RAGE is believed to cause the movement of Aβ across the BBB, resulting in accumulation [39]. In the setting of hyperglycemia, AGE levels also increase RAGE-mediated BBB disruption and potentiate brain pathology [27]. As a result, AGEs have been found in AD brains and the brains of diabetic rats [24, 43]. RAGE also promotes cognitive decline, synaptic impairment, and neurodegeneration, as well as further Aβ production, tangle formation, and tau hyperphosphorylation [27]. This concept has been demonstrated in a mouse AD model, in which downregulation of RAGE signaling in the microglia prevented cognitive impairment and synaptic deficit and reduced stress-related kinase activation [27]. The effects of AGE are further displayed in Fig. 4.
Amyloid deposition

AGE Effects. AGE Effects: Sugars accumulating with age (in T2DM or AD) react with proteins or lipids to generate AGE. AGEs bind to their receptor, RAGE, an interaction associated with effects that culminate in AD hallmarks. Other specific molecules have been thought to also bind to RAGE. Aβ, amyloid-beta; AGE, advanced glycosylation end products; RAGE, advanced glycosylation end-product specific receptor; ROS, reactive oxygen species; SIRT1, sirtuin 1.
AD and T2DM are amyloidogenic diseases. Islet amyloid polypeptide (AIAPP), or amylin, is secreted from the pancreatic β-cells, aggregates to form pancreatic islet amyloid, a feature of T2DM, and deposits in the tissues [35, 44]. AIAPP plays a role in metabolism and glucose homeostasis and is found in over 96%of autopsied diabetics [44, 45]. In AD, cleavage of AβPP by β-site APP cleaving enzyme 1 (BACE-1) and γ-secretase releases Aβ peptides. Aβ can form into different aggregates, such as monomers, oligomers, fibrils, and plaques. Hyperphosphorylated tau also contributes to the formation of NFTs and helical filaments [39]. Aβ and AIAPP share 25%identical amino acid sequences and can cross-seed and deposit in the brain and pancreas [44].
Cognitive decline has been seen when pancreatic AIAPP is found in the brain, and these deposits have been observed in the brain tissue of patients with AD. An analysis of brain samples found that AD patients had 1.4 times higher AIAPP concentrations than non-AD patients [44]. A study also found that higher amyloid concentrations were found in diabetics compared to non-diabetics. APP transgenic mice injected with pancreatic AIAPP aggregates in the brain showed more severe AD pathology and increased memory impairment. AIAPP also accelerated Aβ aggregation in in vitro studies, with fibrils consisting of both peptides [35]. Compared to AIAPP or Aβ alone, AIAPP-Aβ co-oligomerization can increase neuronal cell death up to 3-fold [46].
The gut and microbiota
Dysbiosis, or an imbalance in the gut microbiota, appears to be a factor contributing to many disease states, including AD and T2DM [47]. Research has shown that AD and T2DM patients have decreased levels of bacteria in the phylum Firmicutes. Both disease states also have increased levels in the genus Bacteroides [47]. Various gut microbiota species secrete a mixture of amyloid and lipopolysaccharide, which can polymerize to form insoluble aggregates [48]. This amyloid shares similar properties to cerebral Aβ and can activate inflammatory cells [49]. Antigens from bacteria can move to the CNS, which was demonstrated by increased lipopolysaccharide and Escherichia coli pili in the brains of AD patients, and potentiate an inflammatory response [48].
Trimethylamine N-oxide (TMAO) is an intestinal microbial metabolite associated with several chronic diseases, including T2DM and AD [50]. TMAO causes insulin resistance by stimulating gluconeogenesis and N-nitroso compound formation. N-nitroso compounds contribute to activation of inflammatory pathways, DNA damage, lipid peroxidation, and oxidative stress [50]. A study showed that higher levels of TMAO are associated with diabetes, with the incidence increasing by 54%per 5μmol/L of TMAO [50]. Higher TMAO levels are also correlated with AD and biomarkers of neural degeneration, including phosphorylated tau and neurofilament light chain protein [50]. A study of mice treated with TMAO showed cellular aging in the brain, increased oxidative stress, and reduced cognition [50].
Impaired vasculature and blood-brain barrier
The BBB acts as a barrier that prevents unwanted substances from entering the brain, such as pathogens and plasma proteins, and helps to regulate the chemical composition of the brain [51]. Hyperglycemia promotes oxidative stress and inflammation, which can interfere with the folding of gap junction proteins and astrocyte communication [51]. Breakdown of the BBB due to loss of tight junctions has also been seen in AD [52]. The end-feet of astrocytes normally adhere to the basement membrane of endothelial cells and pericytes of capillaries. In diabetic db/db female mice, it was observed that these astrocyte foot processes were detached in the capillary neurovascular unit [53]. Since astrocytes are important for signal transmission between components of the neurovascular unit, this detachment can cause neuronal dysfunction [53]. The brain endothelial cells of db/db diabetic mice have demonstrated basement membrane thickening and rearrangement [53]. Microglia are also abnormally activated in both diseases, which can cause excessive secretion of inflammatory cytokines and production of oxidative substances [53]. This inflammation and oxidative stress contribute to BBB damage [52].
In AD, decreased cerebral blood flow has been demonstrated, reducing oxygen, glucose, and nutrient supply. Likewise, insulin resistant-states affect PI3K-mediated vasodilation. PI3K stimulates endothelial nitric oxide synthase, which produces nitric oxide and vasodilation. With insulin resistance, vasodilation by PI3K is impaired, decreasing nitric oxide and causing vasoconstriction. This also results in decreased nutrient supply to the brain [14].
In the setting of diabetes, there is decreased oligodendrocyte survival and reduced proliferation, which may cause changes in myelin remodeling and delay in distant information transduction. A decreased number of pericytes and apoptosis has also been noted [53]. In AD, decreased oligodendrocyte differentiation and proliferation has been seen, as well as loss of pericytes [52, 53]. Lipoprotein receptor-related protein 1 (LRP1) regulates brain endothelial cells and helps with clearing Aβ. In T2DM and AD, LRP1 is downregulated, reducing the ability of Aβ to be transported across the BBB for clearance [23, 52].
Genetics
Shared genes and biological pathways
Hu et al. (2020) found that 759 genes were directly or indirectly connected to T2DM and AD, and 2 expression genes, GRMD1B and RP1-111D6.3, are directly connected to T2DM and AD [16]. The study also identified 16 shared pathways between the two diseases [16]. The cyclic adenosine monophosphate (cAMP) response element-binding protein (CREBBP) pathway is responsible for converting short-term memory to long-term memory. Morphine addiction, neuroactive ligand receptor interaction, fatty acid biosynthesis, and primary bile acid biosynthesis pathways are also associated with T2DM and AD [16]. Fatty acids affect neurotransmitter synthesis and signaling and fluidity of cell membranes in the brain; bile acids play a role in cell signaling, met-abolism, inflammation, and immune function. Abnormal bile acids can cause changes in insulin secretion, and fatty acid utilization contributes to insulin resistance. Dopaminergic synapse pathways, the SORCS1 gene, and methylated genes POU3F2, KIF4B, and TNSL3 and are also linked to both diseases [6, 16].
Pereira et al. (2020) identified 17 common bio-markers that are expressed in both AD and T2DM, supporting a link between these disease states: alpha-2-macroglobulin, apolipoprotein A-I, apolipoprotein A-IV, apolipoprotein B-100, apolipoprotein E, ceru-loplasmin, complement C4, galectin-3–binding protein, haptoglobin, Ig α-1 chain C region, Ig μ chain C region, Ig κ chain C region, inter-alpha-trypsin inhibitor heavy chain 1, inter-alpha-trypsin inhibitor heavy chain 2, pancreatic polypeptide, transthyretin, and zinc alpha 2-glycoprotein [54].
Single nucleotide polymorphisms (SNPs)
A genome-wide association study identified 395 SNPs with the same risk allele in T2DM and AD [55]. A meta-analysis identified 8 SNPs shared by both diseases, including TP53INP1 and TOMM40 [55]. T2DM and progression of mild cognitive impairment (MCI) to AD was also associated with SNP rs391300 of the serine racemase gene [55]. The AKT1 rs2498786 CC genotype is associated with insulin resistance and AD [55].
Apolipoprotein E4
The APOE4 allele is a key genetic risk factor for AD, with about 70%of carriers developing the disease [17]. Altered APOE4 is also found in T2DM, with increased risk of AD in diabetic carriers [20, 24]. Carriers of the gene are more likely to develop insulin resistance, as shown by APOE4 mice with impaired glucose metabolism and reduced sensitivity to insulin [17]. Carriers demonstrated peripheral glucose hypometabolism, synaptic dysfunction, decreased cerebral glucose metabolism, reduced Aβ clearance, and neuroinflammation. Higher phosphorylated tau levels in the CSF are also correlated with insulin resistance in APOE4 carriers [27]. APOE4 correlates with increased cerebrovascular Aβ [20]. Although Doney et al. (2019) did not find a formal interaction between T2DM and APOE4, patients with T2DM and APOE4 had a higher risk (HR, 2.83; 95%CI, 2.38-3.36) for AD than patients with only T2DM (HR, 1.42) or APOE4 (HR, 1.8) [56]. While some studies have also found no connection between T2DM and APOE4, others have found that AD pathology was higher in T2DM patients with the APOE4 allele compared to those without [6, 57]. APOE4 tightly binds insulin receptors and prevents their movement to the cell surface. APOE4 also inhibits SIRT1 and suppresses insulin signaling as shown by the lower levels of SIRT1 in APOE4 genotype mice [17].
8-oxoGsn
8-oxoGsn is a product of nucleic acid oxidation that can demonstrate oxidative stress and oxidative RNA damage through tissue, blood, CSF, and urine analysis. Examination of the nucleotides from the brain tissue of AD patients showed increased 8-oxoGsn expression [58]. Another study found that 8-oxoGsn CSF concentrations were 5 times higher in AD patients than controls [58]. Urinary 8-oxoGsn is also a prognostic factor for T2DM, with higher levels corresponding to increased risk of death [58]. Analysis of 8-oxoGsn and urinary 8-oxoGsn in diabetics showed significantly increased levels compared to healthy patients [58].
Vitamins and minerals
Magnesium
Magnesium (Mg) deficits are associated with T2DM and AD. While reduced cellular and/or ionized Mg concentrations and normal serum Mg levels have been seen in T2DM patients, reduced levels of total and ionized serum Mg were found in AD patients [59]. Altered Mg metabolism has been observed in patients with dementia and may impair insulin-mediated glucose uptake. An inverse relationship between Mg intake and incidence of new T2DM cases has been observed [59]. Increased urinary loss of Mg may be a factor in depleted Mg levels, especially considering that Mg excretion is increased by hyperglycemia and hyperinsulinemia. Altered Mg transport may also be caused by insulin resistance [59].
Some studies have shown that Mg supplementation may improve glucose and insulin-sensitivity parameters in T2DM patients and patients at risk of T2DM, but trials in this population are limited [59]. Since Mg plays a role in neuronal maturation and cell membrane stability and integrity, it may also have a protective effect in AD. Potential actions of Mg include reducing neuroinflammation, inhibiting abnormal tau protein phosphorylation, inhibiting AβPP processing, allowing toxin clearance, and rev-ersing deregulation of NMDA receptors. In AD mod-els, Mg-L-threonate reduced neuroinflammation, decreased Aβ deposition, and improved learning and memory [59]. Animal studies also suggest that Mg supplementation can slow memory and cognitive decline if started early enough. Although some human studies have found that Mg-rich diets can reduce the risk of cognitive impairment, long-term trials are needed [59].
Calcium
Dysregulation of calcium homeostasis is thought to be a shared factor in T2DM and AD [60]. Production of free radicals causes calcium elevations, which can impact mitochondrial processes and lead to oxidative stress [60]. In T2DM and AD, intracellular calcium is chronically elevated, causing Src non-receptor tyrosine kinase and p52Shc adaptor protein to gather at the membrane surface. In NIH3T3 cells stimulated with saturated fatty acids, this causes JNK activation. Together, Src and JNK cause hyperphosphorylation of IRS-1 and reduced AKT activity. JNK activation is also associated with hyperphosphorylation of tau and neuronal loss [61]. Increased free intracellular calcium reduces levels of Ca2 + /calmodulin-dependent protein kinase II (CaMKII). The binding of CaMKII and CREB is involved in cognition; therefore, disrupted calcium signaling can impede this process. Impairment in calcium signaling can result in neuronal death and cognitive impairment [61].
POTENTIAL THERAPIES
With continued research and findings of the pot-ential mechanisms of AD and their links to T2DM, more treatment options targeting the mutual pathological mechanisms have been identified. There have been studies on repurposing known T2DM agents, such as GLP-1 receptor agonists, dipeptidyl pep-tidase-4 (DPP-4) inhibitors, peroxisome prolife-rator-activated receptor-gamma (PPARγ) agonists, metformin, sodium-glucose co-transporter 2 (SGLT2) inhibitors, and intranasal insulin for AD patients. Additionally, molecules with novel mechanisms of counteracting the progression of AD are in develo-pment with some reaching phase 3. Beyond these treatment options, it is important to note non-pharmacologic interventions that may improve cognitive function in patients with AD.
GLP-1 receptor agonists
GLP is an incretin hormone secreted in the distal small intestine in response to food entering the duodenum [62]. GLP-1 receptors are found in the pancreas, vascular endothelium, and CNS (hypothalamus, cerebral cortex, hippocampus, and olfactory bulb) [63]. When GLP-1 binds to its receptor, it stimulates insulin secretion, blocks glucagon secretion, decreases appetite, and delays gastric emptying [62]. GLP-1 receptor is also involved in regulation of neuronal plasticity and cell survival [64]. GLP-1 receptor agonists have been effective in treating diabetes and obesity, but since AD mouse models and human AD brains show reduced GLP-1 and GLP-1 receptors, their use is of interest in treating patients with AD [65].
Several studies have demonstrated the effect of GLP-1 agonism in mouse models and its potential benefit in AD. Holubov
As preclinical evidence alludes to neuroprotective effects that GLP-1 agonism can provide in AD, recent clinical trials have taken place to solidify the clinical significance of liraglutide. In a double-blind placebo-controlled study, neural activities of liraglutide were evaluated in middle-aged patients (half of the patients with family history of AD) expressing subjective symptoms suggestive of cognitive decline [69]. Patients in the study were randomly assigned to liraglutide or placebo and received treatment for 12 weeks. This study found that higher insulin resistance (measured by fasting plasma glucose levels) led to decreased connectivity between the bilateral hippocampus and anterior medial frontal areas [69]. In addition, patients in the liraglutide arm noticed improvement in this intrinsic connectivity within the default mode network. The results of this study encouraged larger trials of longer duration, such as the ELAD (Evaluating Liraglutide in Alzheimer’s Disease) study, to be conducted in order to determine the benefits of liraglutide in human subjects with AD [69, 70]. The ELAD study was a phase 2b, multicenter, randomized, double-blind, placebo-controlled trial evaluating the effect of liraglutide compared to placebo in patients with mild AD for 12 months [70]. Preliminary results were presented at the 2020 Clinical Trials on Alzheimer’s Disease (CTAD) conference and showed that liraglutide did have a significant effect on the primary endpoint of cerebral glucose metabolic rate in the cortical regions, as no difference in glucose uptake was registered between treatment and placebo groups [71]. However, liraglutide improved three of five secondary endpoints, including better Alzheimer’s Disease Assessment Scale (ADAS)-Exec score and less loss of temporal lobe and total brain gray matter volume [71]. There were no major safety concerns in the study; fewer serious adverse events and no deaths were seen with liraglutide compared to the placebo [71].
Several other GLP-1 receptor agonists have also been evaluated. An et al. (2019) evaluated exenatide in 5-month-old male 5xFAD mice and found improved learning ability and spatial memory [72]. Exenatide also reduced amyloid plaques and improved synaptic degradation. Compared to the AD group, the AD plus exenatide group had more normal synapses, more synaptic vesicles, reduced synaptic cleft width, thickened postsynaptic density, and increased synaptic marker proteins [72]. Mitochondrial dynamics were normalized, along with a significant increase in mitochondrial surface area and decrease in oxidative stress [72]. Conversely, another study of exenatide found no differences compared to placebo in cognitive measures, cortical volume and thickness, and CSF, plasma, and plasma neuronal extracellular vesicle biomarkers [73]. Despite this, a reduction in Aβ42 in extracellular vesicles was observed [73]. Exenatide was well-tolerated, although nausea occurred at a significantly higher rate in the treatment group compared to the placebo group [73].
A study of AD-induced mice intracerebroventricularly administered streptozocin showed that dulaglutide reduced escape latency in the Morris water maze (MWM) and helped with learning and memory [74]. Associative learning was enhanced, and phosphorylation of tau and NFTs was reduced. Significantly increased GLP-1 and GLP-1 receptor levels were also observed [74]. Similarly, a study of lixisenatide in 12-month-old APP/PS1/tau female mice demonstrated improved memory deficits, spatial learning, and long-term potentiation [64]. Lixisenatide significantly lowered inflammation and the percentages of hippocampal Aβ-area, phosphorylated tau-positive cell numbers, and phosphorylated tau-area [64]. Improved cognition has also been demonstrated by GLP-1/glucose-dependent insulinotropic polypeptide (GIP) dual agonists and GLP-1/GIP/glucagon receptor agonists [75–77].
DPP-4 inhibitors
As mentioned previously, GLP-1 is essential for insulin secretion. However, DPP-4 works by rapidly degrading GLP-1, blocking its insulinotropic effects. By inhibiting DPP-4, degradation of GLP-1 is blocked, enhancing the levels of GLP-1 and GLP-1R and helping with insulin resistance [65]. Since GLP-1 has shown benefit in AD, research evaluating multiple DPP-4 inhibitors has been conducted to evaluate their efficacy in treating the disease.
To determine the effect of DPP-4 inhibitors on AD-like neurodegeneration, sitagliptin and saxagliptin were given to 10-month-old AD mice for 8 weeks. Results showed an increase in GLP-1 and GLP-1 receptor level and a significant reduction in Aβ1 - 42 levels in the cortex and hippocampus of AD mice [65]. Results of the MWM test showed that AD mice treated with sitagliptin and saxagliptin had enhanced spatial learning and memory. Both medications also improved synaptic structure by increasing synaptic protein expression in the brain [65]. Sitagliptin and saxagliptin reduced hyperphosphorylated neurofilaments, decreased O-GlcNAcylation, and improved GLP-1 signal transduction [65]. Comparably, a study of sitagliptin given to APP/PS1 mice for 8 weeks showed significantly reduced amyloid plaque deposition and increased dendritic spine density [78]. In the MWM, mice had improved spatial learning and cognition. Increased treatment benefit was seen in the 7-month-old mice compared to the 9-month-old mice, suggesting that sitagliptin may be better for early stage-AD [78]. Clinical evidence of sitagliptin’s benefit in AD was demonstrated by improved Mini-Mental State Examination (MMSE) scores in diabetic patients with AD treated for 6 months [79].
Ma et al. (2018) found that vildagliptin reduced escape latency and improved memory impairment and spatial learning in the MWM test [80]. Vildagliptin downregulated AβPP and phosphorylated tau expression, increased phosphorylated AKT, decreased phosphorylated GSK3β, and promoted expression of synaptic plasticity-associated proteins (PSD-95 and synaptophysin) [80]. In vivo studies of vildagliptin and saxagliptin in rats have also shown a reduction in Aβ42 and total tau levels in the hippocampus and suppression of neuroinflammation [79]. Similar results were seen with linagliptin in a mouse AD model, as well as inhibition of cognitive decline and decreased tau phosphorylation [79, 81]. Overall, DPP-4 inhibitors have displayed beneficial effects on neurodegeneration by suppressing oxidative stress, Aβ plaque formation, tau phosphorylation, and neuroinflammation [81]. A significant association between DPP-4 inhibitors and protection against deterioration in cognitive function has been supported by clinical research [79].
PPARγ agonists
PPARγ agonists, or thiazolidinediones, are used to treat T2DM by acting on nuclear receptor and the transcription factor PPARγ to produce gene expressions for improved glucose metabolism and insulin resistance [82]. Although the main mode of action is to reduce brain insulin resistance by enhancing insulin receptor sensitivity, PPARγ agonists target multiple AD pathophysiological factors, including phosphorylated tau, Aβ levels, microglial function, inflammation, mitochondria, lipid homeostasis, cell death and brain atrophy, and cerebral glucose metabolism [83].
Preclinical studies delved into the potential utilization of rosiglitazone in AD by looking at mouse models [82]. A 2020 study provided genomic and proteomic data that revealed a potential association between PPARγ and mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinase (ERK) signaling in the hippocampus, the memory center of the brain. More specifically, the relation between PPARγ and phospho-ERK network was discovered, which could signify a positive effect of PPARγ agonists on memory consolidation. The Tg2576 mouse model was used in this study, which demonstrated glucose dysregulation and insulin resistance, overproducing Aβ and leading to memory impairment commonly seen in human late-onset AD. The 8-month-old mice were given rosiglitazone for a month to determine whether this PPARγ agonist could bring about positive effect on cognitive modalities, such as spatial navigation, context discrimination, and object recognition learning and memory associated with different parts of the hippocampus. Some methods implemented included insulin sensitizer treatment, MWM, novel object recognition, foreground fear conditioning, and context discrimination. The results showed that rosiglitazone treatment led to improvements in select areas of the hippocampus, predominantly the dorsal hippocampus (learning and memory in MWM and background fear conditioning) rather than the ventral hippocampus (context discrimination fear conditioning) [82]. Other studies of rosiglitazone corroborated these findings by showing an association between treatment and improved memory and reduced learning deficits [83]. These improvements correlated with enhanced short-term plasticity and synaptic activity. Reduced Aβ40, Aβ42, and phosphorylated tau were also observed [83]. In a phase 2 clinical trial of patients with early AD and amnestic MCI, rosiglitazone treatment for 24 weeks was statistically associated with improved delayed recall at 4 and 6 months. Conversely, another trial of rosiglitazone in patients with mild to moderate AD showed no sustained effect on brain volume and glucose metabolic rate. There was also no effect on ADAS-Cognitive Subscale (ADAS-Cog) or clinician’s interview-based impression of change with caregiver input [83]. This discrepancy was further noted in two phase III studies, where no statistically or clinically significant effects in patients with mild to moderate AD were seen.
Multiple studies have also evaluated the use of pioglitazone in AD. Combination treatment with pioglitazone and leptin in an AD mouse model showed improved spatial memory and reduction of synapse loss, neocortical glial response, and Aβ deposits [84]. Several preclinical studies have found that pioglitazone diminished amyloid plaque deposits, hyperphosphorylated tau, reactive mic-roglia and astrocytes, and Aβ40 and Aβ42. Memory retention, long-term potentiation, and cerebral blood flow and glucose uptake were improved [83]. A clinical trial of 24-week pioglitazone treatment in mild AD demonstrated statistically improved MMSE, Japanese ADAS-Cog (ADAS-JCog), and revised Wechsler Memory Scale (WMS-R). On the other hand, a longer 72-week trial found that pioglitazone was not statistically correlated with improved cognition [83]. Variable results regarding the efficacy of PPARγ agonists necessitate further research.
Metformin
Metformin is considered a first-line medication for the treatment of T2DM. It works by increasing uptake of glucose through activation of AMP-activated protein kinase (AMPK). Metformin has shown reduction in inflammation, oxidative stress, and the formation of AGEs [85]. A study of metfor-min in APP/PS1 female mice demonstrated shortened escape latency in the MWM, indicating improvement in spatial learning [86]. Metformin-treated mice had reduced neuronal apoptosis and increased hippocampal neurogenesis. Metformin significantly reduced the density of Aβ40-positive and Aβ42-positive plaques and levels of soluble Aβ40 and Aβ42. Fewer reactive microglia and astrocytes were detected around Aβ plaques in metformin-treated mice, and IL-1β and TNF-α were drastically decreased. Furthermore, metformin promoted anti-inflammatory IL-4 in the hippocampus and cortex [86].
A meta-analysis of 23 studies showed that metformin use led to a lower likelihood of developing cognitive impairment (OR, 0.55; 95%CI, 0.38–0.78) [85]. Similar results were found in an analysis of patients classified as having dementia by the MMSE. Another meta-analysis comparing diabetics who received metformin to those who did not showed that use of metformin decreased the risk of developing dementia or AD (HR, 0.76; 95%CI, 0.60–0.97; p = 0.03) [85]. Conversely, in a cohort of patients who were cognitively intact, had MCI, or had AD, metformin was associated with decreased cognitive performance [85]. Other studies have also found that metformin worsens cognition in AD or fails to provide benefit [87, 88]. Conflicting results regarding the utility of metformin warrant further study.
SGLT2 inhibitors
SGLT2 inhibitors are used to treat diabetes by lowering glucose levels through prevention of tubular glucose reabsorption [89]. Significant SGLT2 expression has also been identified in the cerebellum, hippocampus, endothelial cells, and BBB. Limited research has assessed the effects of SGLT2 inhibitors in AD, but some reports suggest possible neuroprotection [90]. Empagliflozin was evaluated in a mixed murine AD and T2DM model (APP/PS1xdb/db). Use of empagliflozin demonstrated a significant improvement in the new object discrimination test, reduced acquisition time in the MWM, and improved impairment in the retention phase, although there were no significant differences between groups [89]. Although a statistically significant reduction in the hippocampus was not reached, empagliflozin treatment decreased tau phosphorylation and brain atrophy in the cortex. Microglial burden in the parenchyma was also reduced [89]. So far, no preclinical and powered clinical research has provided enough evidence for the benefits of SGLT2 inhibitors in AD [91].
Intranasal insulin
Intranasal insulin works by bypassing the BBB, allowing insulin to reach the hippocampus and cortex [63]. Since insulin resistance seems to be a mechanism involved in AD, delivering insulin into the CNS is an appealing treatment. Small trials of patients with early AD and MCI showed improved recall in APOE4-negative patients, but deterioration in APOE4 carriers [92]. The first multi-site phase II/III trial of intranasal insulin in patients with AD and MCI involved 240 patients and evaluated the feasibility, safety, and efficacy of treatment; 121 and 119 patients were randomized to the insulin and placebo groups, respectively. The primary outcome was mean score change in ADAS-cog-12. Secondary outcomes included Alzheimer Disease Cooperative Study Activities of Daily Living Scale for Mild Cognitive Impairment (ADL-MCI) and Clinical Dementia Rating Scale Sum of Boxes (CDR-SB). No differences were reported in ADAS-cog-12 or performance on the CDR-SB, ADL-MCI, or memory composite tests [93]. No differences were found in CSF Aβ42 and Aβ40, total tau, tau p-181, or Aβ42:Aβ40 or Aβ42:total tau ratios. Additionally, CSF insulin levels, blood glucose, and hemoglobin A1C remained unchanged, and small but significant decreases in hippocampal volume were seen in the insulin group [93]. The most frequent adverse events were infections, respiratory disorders, nervous system disorders, and injuries. A higher but insignificant rate of vascular disorders occurred in the insulin group [93].
New targets for treating AD
With continued research efforts and findings, AD is now understood to have features of a metabolic disorder. Thus far, studies have been conducted to explain several mechanisms of metabolic dysfunction, such as glucose metabolism, adipose tissue, mitochondria, and metabolic correlation to neuroinflammation and neurodegeneration [94]. As a result of these findings, new approaches in treating AD have emerged. New AD biomarkers are being discovered; currently, there are 136 active trials investigating 121 different molecules for treating AD. Molecules focusing on immunotherapy to inhibit the build-up of extracellular Aβ plaques and improve synaptic plasticity and neuroprotection are in the late clinical phase. Table 1 highlights small molecules in phase III trials.
Small Molecules Targeting Alzheimer’s Disease in Phase III Trials [94]
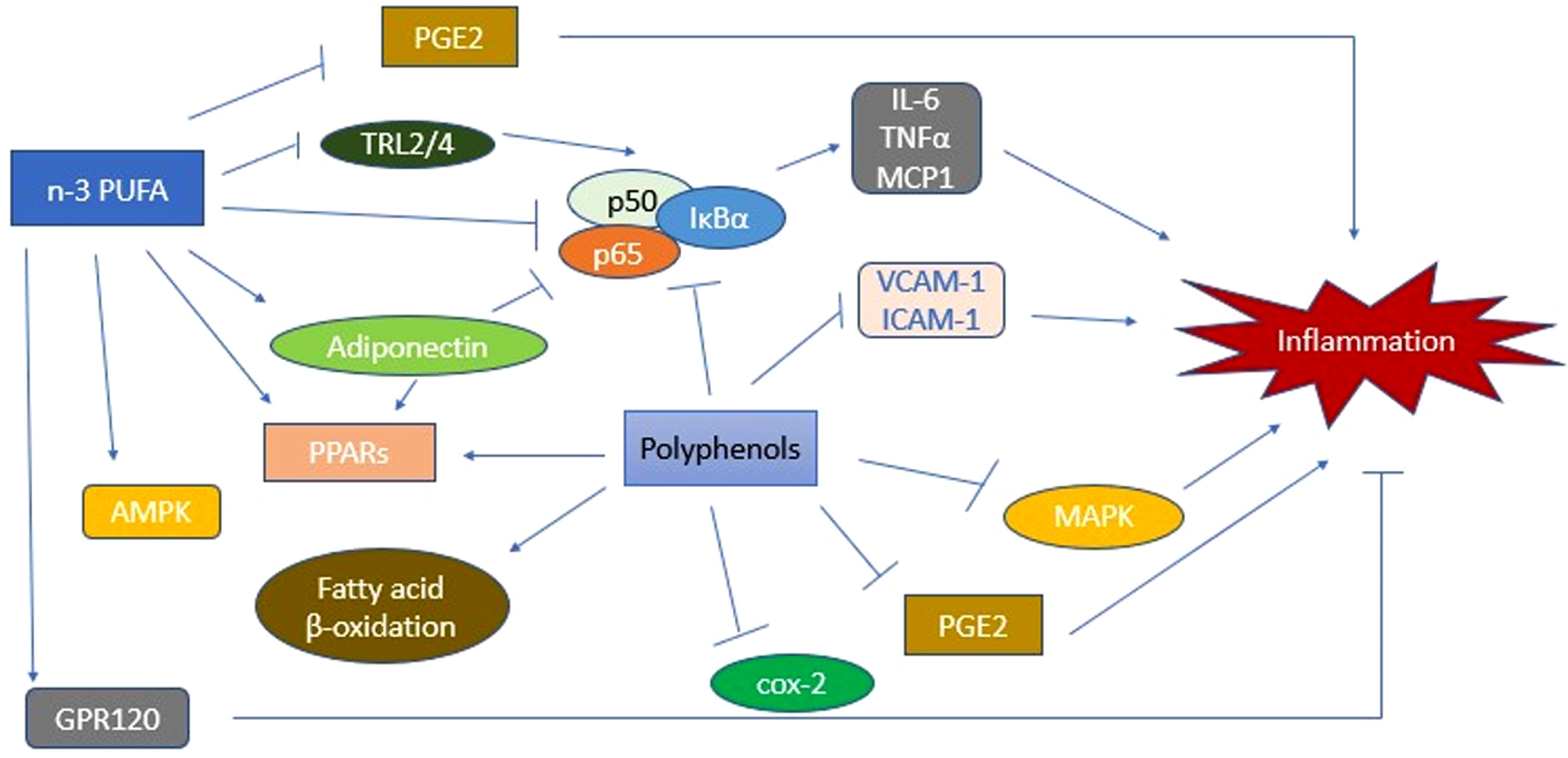
Effect of PUFAs on the Inflammatory Pathway. PUFAs initiate a cascade of activities that lead to inhibition of various inflammatory mechanisms. AMPK, AMP-activated protein kinase; cox-2, cyclooxygenase-2; ICAM-1, intercellular adhesion molecule 1; IL-6, interleukin 6; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemoattractant protein-1; PGE2, Prostaglandin E2; PPARs, peroxisome proliferator-activated receptors; PUFA, polyunsaturated fatty acids; TNF-α, tumor necrosis factor alpha; TRL2/4. Toll-like receptor 2/4; VCAM-1, vascular cell adhesion molecule 1.
In addition to small molecules, monoclonal anti-bodies targeting Aβ and tau oligomers are in development, including Aducanumab (targeting plaques and oligomers), BAN2401 (targeting protofibrils), Gantenerumab (targeting plaques and oligomers), and Solanezumab (targeting monomers) [94]. Vaccination therapy (CAD106; Novartis) has also been considered in the past and was in phase II/III [94]. However, in September 2019, the trial was retired. Another approach in treating AD lies in the microRNAs and their regulatory potential. Antisense oligonucleotides (ASOs) are being studied with the purpose of targeting GSK3β and tau [94]. In mouse models, ASOs have shown improvement in memory and learning, while decreasing oxidative stress and pathological tau build-up [94]. The mechanisms of AD etiology will continue to be studied, and new and old therapies will continue to influence the treatment approaches to AD.
Non-pharmacologic interventions
As we delve further into non-communicable diseases such as AD, it has been solidified that there are common biochemical mechanisms such as chronic impairment of inflammatory pathways and redox dysregulation. With further understanding of the etiology of non-communicable diseases, lifestyle risk factors are continually being identified, and non-pharmacologic treatment and prevention approaches are being studied. Various research efforts have studied dietary components of inflammatory processes and measures to reduce the low-grade inflammation that may be linked to non-communicable diseases including T2DM and AD. Recommended interventions to prevent such inflammatory response include a high-fiber, low-carbohydrate diet coupled with exercise [95]. As inflammation is concluded to be a major factor leading to the development of T2DM and AD, research has identified the role of several nutrients in the inflammatory pathway schema. For instance, studies have identified the benefits of polyunsaturated fatty acids (PUFAs) in reducing inflammation [95]. The mechanism relating PUFAs to inflammation is shown in Fig. 5. PUFAs can be acquired via consumption of plant-based seeds, nuts, and fatty fish [95]. Another nutrient that has been shown effective in mitigating the risk for AD linked with T2DM is resveratrol, a stilbenoid that could permeate into the brain. Found in fruits, such as blueberries, mulberries, raspberries, and grapes, resveratrol has neuroprotective effects proven in rat models [96]. Limited clinical trial experience with resveratrol has shown improved neurovascular coupling capacity and ability to multitask [96]. Curcumin, a component of the spice tumeric, has shown anti-amyloid and antioxidant effects, reduction in systemic inflammation, and improved memory functions [97, 98]. In mouse models of AD, curcumin has demonstrated improvement in cognitive decline and synaptic function, as well as amelioration of defective insulin signaling [97]. Although further investigation is warranted, these findings show that dietary nutrients and lifestyle factors play an important role in regulating signaling pathways and subsequently improving immune function and optimizing metabolic homeostasis in patients with metabolic diseases including T2DM and AD [96].
CONCLUSION
In this review, we have presented the recent discoveries of potential mechanisms of AD pathogenesis in relation to that of T2DM. The science of AD pathology is constantly being updated, and the association between AD and T2DM, as well as other noncommunicable diseases, is becoming clearer. With these findings, AD is being understood more as a metabolic disease.
Furthermore, we highlighted the potential bio-markers that could be utilized to more effectively target AD and introduced a new treatment paradigm to target the mechanisms that may cause AD and its progression. With the understanding of AD as a metabolic disease, similarities in disease mechanisms are constantly being discovered. Insulin signaling is a main component of T2DM pathology. In this review, we detailed how insulin resistance may also potentiate neurodegeneration and cognitive impairment leading to AD. This causative relationship of insulin resistance and AD has alluded to its rising title, “type 3 diabetes mellitus.” Moreover, we detailed other mechanisms correlating to the disease mechanisms in T2DM. Dysregulation of insulin receptors and various components of the insulin signaling pathway, including AKT, GSK3β, and mTOR, are shared in both diseases. T2DM and AD also show evidence of inflammation, oxidative stress, production of AGEs, and amyloid deposition. The impact that other factors, such as mitochondrial dysfunction, changes in neurovascular structure, and genetics, have on the development of these conditions is also being explored.
With the discovery of factors contributing to AD, innovative approaches to manage AD are in place and currently in development. Investigators are evaluating the efficacy of T2DM medications, such as GLP-1 receptor agonists, DPP-4 inhibitors, PPARγ agonists, metformin, SGLT2 inhibitors, and intranasal insulin in AD. Furthermore, there are 136 active trials at different stages involving 121 therapeutic agents targeting novel AD biomarkers. Further understanding of AD etiology has also elucidated lifestyle risk factors for AD, and non-pharmacological prevention and therapies have been introduced. Moving forward, the mechanisms of AD etiology will continue to be studied, and new and old therapies will continue to influence the treatment approaches to AD, getting one step closer to alleviating its impact.