
Abstract
The haploinsufficiency of the methyl-binding domain protein 5 (MBD5) gene has been identified as the determinant cause of the neuropsychiatric disorders grouped under the name MBD5-neurodevelopment disorders (MAND). MAND includes patients with intellectual disability, behavioral problems, and seizures with a static clinical course. However, a few reports have suggested regression. We describe a non-intellectually disabled female, with previous epilepsy and personality disorder, who developed early-onset dementia. The extensive etiologic study revealed a heterozygous nonsense de novo pathogenic variant in the MBD5 gene. This finding could support including the MBD5 gene in the study of patients with atypical early-onset dementia.
Keywords
INTRODUCTION
The 2q23.1 microdeletion syndrome was described in patients with moderate to severe intellectual disability, language impairment, behavioral difficulties, seizures, ataxia, microcephaly, and other dysmorphic features [1–4]. In 2011, after analyzing a large cohort of patients with genetic alterations of chromosomal region 2q23.1, the haploinsufficiency of the methyl CpG-binding domain 5 gene (MBD5) was identified as the mechanism and the gene responsible for the disorder [4]. Pathogenic variants in the MBD5 gene produce a similar but typically milder clinical phenotype [4]. In some cases, inherited variants with different degrees of severity have been reported among family members, suggesting incomplete penetrance and variable expressivity [5, 6]. MBD5-Associated Neurodevelopment Disorders (MAND; #156200) was the term proposed to describe the syndromes associated with this altered gene, including deletion, duplications, and variants [5].
The MBD5 gene encodes a protein expressed throughout the brain and localized in non-heterochromatin regions of the nucleus, suggesting that MBD5 acts regulating gene transcription [7, 8].
In MAND, the central phenotype shares a common set of neurodevelopmental, cognitive, and behavioral impairments with a static clinical course [5]. However, a few cases of regression have been reported [9–12]. Here, we describe a heterozygous nonsense variant in the MBD5 gene in a 48-year-old woman with early-onset dementia. Additionally, we review the literature on cognitive and behavioral regression associated with MBD5 haploinsufficiency.
MATERIALS AND METHODS
Patient data were obtained from the medical records of the Hospital Universitario 12 de Octubre in Madrid (Spain). This study has been approved by the ethical committee/institutional review board. The patient’s father provided written informed consent.
DNA analysis and sequencing
Whole exome sequencing (WES) enrichment and libraries were prepared using the xGen Exome Panel v1.0 kit (Integrated DNA Technologies, Coralville, IA, USA). Paired-end sequencing (2×75 bp) was carried out on a NextSeq 550 NGS sequencer (Illumina, San Diego, CA, USA). A validated custom pipeline, KarMa, was used for the bioinformatics analysis. WES data were prioritized using the following Human Phenotype Ontology (HPO) terms: Personality disorder (HP:0012075), Behavioral abnormality (HP:000708); Depression (HP: 000716); Dementia (HP:0000726); Supranuclear gaze palsy (HP:0000605); and Seizures (HP:0001250). The variant classification was done according to ACMG criteria [13]. Segregation analysis was performed by Sanger sequencing, following manufacturers’ instructions.
RESULTS
Our patient is a 48-year-old right-handed woman with no relevant neurological or psychiatric disorders on family history. Pregnancy, birth, and early development were unremarkable. At 28 months of age, she suffered a non-febrile convulsive seizure. She studied elementary, high school, and a college degree without problems. The patient was evaluated in psychiatric consultations at 23 years because of emotional instability and impulsivity problems. Later, obsessions and compulsions with cleaning and auto- and hetero-aggressiveness were added, being diagnosed with a mixed personality disorder. She also suffered from anhedonia, depressed mood, and suicidal ideations. In the following years, the patient showed behavioral fluctuations, interpersonal relationship difficulties, and short periods of alcohol abuse while living with her parents. She kept on follow-up by Psychology and Psychiatry with the diagnosis of mixed personality disorder. She worked as a sales promoter, a travel agent, and a television figurant in those years. From 30 to 46 years, she had several psychiatric admissions related to suicidal attempts and disruptive behavior. Impulsivity, empathy, and feelings of guilt and shame were common during those years.
At the age of 39, the patient was evaluated by a neurologist to diagnose unknown onset tonic-clonic seizures. The neurological examination, electroencephalography (EEG), and magnetic resonance imaging (MRI) studies were unremarkable. During the follow-up, several recordings were made, but only a sleep-deprived EEG test showed interictal epileptic discharges in the left frontal region, supporting non-lesional focal epilepsy diagnosis. During the following years, her seizures were well-controlled with lacosamide and lamotrigine without adverse effects.
When she was 46 years old, her parents described fluctuating speech problems with difficulties in expression and finding words. In a neurocognitive assessment (Table 1), she was slightly inattentive and showed mild dysnomia. She could repeat sentences and understanding sequential orders without problems. She recalled recent events as the food of the previous day, and praxis function was preserved. Her Mini-Mental State Examination (Folstein) score was 29/30, and she scored 10 in the semantic verbal fluency test (animals) and 15/18 in the Frontal Assessment Battery. The rest neurological examination was unremarkable. Complete blood tests to help diagnose dementia and rule out other conditions were all within normal limits. A new brain MRI was normal. Over the next few months, the patient presented a behavioral and cognitive worsening with more impulsivity and aggressiveness, cessation of self-care, and loss of ability to handle her medication. At that moment, she was institutionalized in a psychiatric center, achieving an improvement in her behavior, although without recovering her previous cognitive and functional situation.
Neuropsychological assessment of the patient
Pc, Standard punctuation; Percentile (normative data adjusted by age and sex in Spanish population). WAIS-III, Wechsler Adult Intelligence Scale; WMS, Wechsler Scale Memory; N/A, not available. *Total score: correct answer. ∧Total score: time in seconds.
A new neurological assessment at 48 years old confirmed a frontal-dysexecutive cognitive decline, and a mild dementia diagnosis was established. She had an upgaze restriction with a “round the houses” sign. Her gait was slightly wide-based. A complete neurological examination showed no other abnormalities. An extensive laboratory workup, including cerebrospinal fluid analysis with Alzheimer’s disease biomarkers (Aβ40, Aβ42, 42/40 index, total-tau, and phosphorylated-tau), neural-specific, and onco-neural antibodies, and repeated MRI, revealed no abnormalities. The EEG showed diffuse, mild, and poor reactivity slowing with non-epileptiform abnormalities, and the brain 18F fluorodeoxyglucose-positron emission tomography displayed hypometabolism in bilateral parietotemporal and frontal regions (Fig. 1).
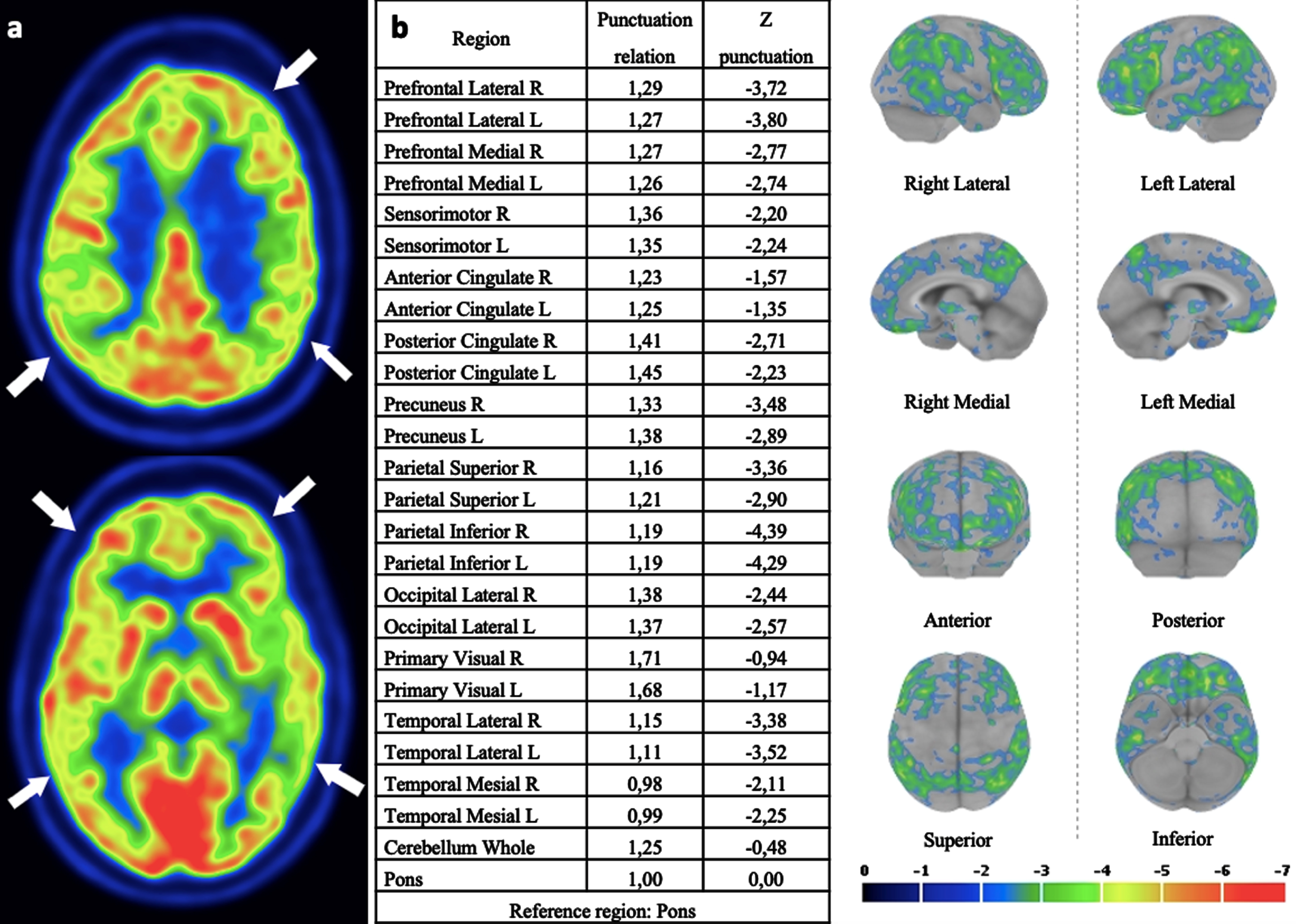
18F-FDG PET/CT images of the patient (a). Statistical parametric maps showing decreased metabolism in bilateral parietotemporal and frontal regions, using dedicated software Cortex ID Suite, GE Healthcare software (b).
In summary, the patient presented a complex medical history of long-term psychiatric symptoms, epilepsy, mild oculomotor abnormalities, and dementia with frontal features in the last two years. A heredodegenerative cause was suspected, and directed genetic studies were performed. The prioritized WES analysis revealed the presence of the heterozygous nonsense c.973C>T p.(Arg325Ter) variant in the MBD5 (NM_018328.4) (*611742) gene, a variant not reported in the consulted population databases (gnomAD, 1000 G, Kaviar) and classified as pathogenic in ClinVar (ID 521004). The study for genes related to genetically determined dementia and disorders that could explain the patient’s phenotype, such as MAPT, NPC1, NPC2, TARDBP, etc., was negative. Segregation analysis showed that none of her parents were heterozygous carriers, confirming a de novo origin of the pathogenic variant in the patient.
DISCUSSION
Here, we report a de novo pathogenic nonsense MBD5 variant in heterozygosis associated with early-onset dementia on a case emerging in early adulthood with mixed personality disorder and seizures. At the age of 48 years, after two years of progressive behavioral changes reflecting frontal lobe dysfunction and language problems, the diagnosis of dementia was established, which was consistent with bilateral frontotemporal and parietal lobe hypometabolism in the FDG-PET image.
Neurodevelopmental regression has been reported in a few pediatric cases with intellectual disability due to MBD5 deletion [9, 10], but only two case reports in the literature have described a relationship of MBD5 mutations with adult-onset dementia (Table 2). A 60-year-old woman with severely intellectually disabled autism and refractory epilepsy presented a cognitive and behavioral decline in her fifties. A WES and subsequent analysis of variants within a panel of known intellectual disability disease genes disclosed a heterozygous pathogenic frameshift mutation of MBD5. Compared with this patient, our proband showed no previous intellectual disability and well-controlled epilepsy. The other was a 44-year-old man with bipolar and anxious disorder and a microdeletion involving at least five genes, including MBD5, who developed early-onset dementia without more clinical details given [11].
Characteristics of patients with MBD5 haploinsufficiency and early-onset dementia
Our case also expands the phenotypic spectrum of MBD5 mutations to include a clinical phenotype (normal development until early adulthood, with psychiatric manifestation and seizures preceding dementia and supranuclear gaze palsy) resembling late-onset Niemann-Pick type C disease [14].
Despite the evidence that disruption of MBD5 adversely affects neurodevelopment and proper neuronal function, little is known about the altered biological processes [8, 15]. Recently, the transcriptional landscape of MBD5 haploinsufficiency across multiple brain regions of a heterozygous Mbd5 + /GT mouse model has been described. Genes displaying increased or reduced expression showed subtle changes, with the cortex as the most affected region. Furthermore, the transcriptional consequences of MBD5 disruption in CRISPR-derived neurons showed no meaningful overlap between gene expression and the animal model, so that MBD5 deficiency can have quite different effects depending on cell type and cell state [16]. Indeed, genes important in neurodevelopment are expressed in early developmental and adult human brains [17]. Therefore, we cannot speculate about molecular mechanisms responsible for dementia in this case, but studies evaluating cumulative or long-term effects induced by gene expression dysregulation by MBD5 during mouse lifespan neurodevelopment could shed light on it.
In conclusion, we present a de novo nonsense MBD5 pathogenic variant in a patient with no previous intellectual disability and early-onset dementia. The expansion of the new genetic techniques in daily clinical practice identifies new genes causative of neurological disorders and new phenotypes associated with their mutations. In this context, phenotype-driven prioritization of candidate genes facilitates the differential diagnosis process improving our genetic diagnosis yield. It is a useful and recommended approach towards genomic diagnostics in rare diseases [18]. Although there are only a few case reports, the genetic study of MBD5 could be considered in adult patients with early-onset cognitive impairment of unknown origin, even in cases without previous intellectual disability.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0648r1).