
Abstract
Background:
Alzheimer’s disease (AD) is the most common form of dementia in older adults and extracellular accumulation of amyloid-β (Aβ) is one of the two characterized pathologies of AD. Obesity is significantly associated with AD developing factors. Several studies have reported that high fat diet (HFD) influenced Aβ accumulation and cognitive performance during AD pathology. However, the underlying neurobiological mechanisms have not yet been elucidated.
Objective:
The objective of this study was to explore the underlying neurobiological mechanisms of HFD influenced Aβ accumulation and cognitive performance during AD pathology.
Methods:
2.5-month-old male APP/PS1 mice were randomly separated into two groups: 1) the normal diet (ND) group, fed a standard diet (10 kcal%fat); and 2) the HFD group, fed a high fat diet (40 kcal%fat, D12492; Research Diets). After 4 months of HFD or ND feeding, mice in the two groups were subjected for further ethological, morphological, and biochemical analyses.
Results:
A long-term HFD diet significantly increased perirenal fat and impaired dendritic integrity and aggravated neurodegeneration, and augmented learning and memory deficits in APP/PS1 mice. Furthermore, the HFD increased beta amyloid cleaving enzyme 1 (BACE1) dephosphorylation and SUMOylation, resulting in enhanced enzyme activity and stability, which exacerbated the deposition of amyloid plaques.
Conclusion:
Our study demonstrates that long-term HFD consumption aggravates amyloid-β accumulation and cognitive impairments, and that modifiable lifestyle factors, such as obesity, can induce BACE1 post-modifications which may contribute to AD pathogenesis.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia, which seriously threatens human health [1]. The main clinicopathologic features of AD are amyloid-β (Aβ) plaques and neurofibrillary tangles within the brain. Before the neurofibrillary tangles aggregate with hyperphosphorylated tau, Aβ plaques are already detectable in the brain [2]. Multiple studies have also demonstrated that Aβ oligomers directly induce tau hyperphosphorylation and results in neurotoxicity affecting synaptic plasticity and cognitive performance [3–5]. Aβ peptides are derived from the sequential proteolysis of the amyloid-β protein precursor (AβPP) [6]. Because the cleavage site of α-secretase occurs in the middle of the Aβ domain, the activation of this enzyme precludes the generation of Aβ [7]. In amyloidogenic proteolytic processing, AβPP is initially cleaved by beta amyloid cleaving enzyme 1 (BACE1), which generates AβPPβ and C-terminal fragment C99 [6, 8]. C99 is further cleaved by the γ-secretase complex to produce Aβ [9]. Therefore, BACE1 may be the rate-limiting enzyme in the amyloidogenic process [8].
BACE1 is expressed at high levels in neurons, astrocytes, and oligodendrocytes, and is particularly abundant in neurons [10, 11]. After translation, BACE1 predominantly resides in the endoplasmic reticulum and the trans-Golgi network (TGN) of the Golgi apparatus for protein modification and folding [12–14]. After post-synthesis processing, BACE1 can be transported to the plasma membrane and then endocytosed into early and late endosomes [15, 16]. From here, BACE1 can be reverse-transported to the TGN for recycling [17]. Because an acidic environment is optimal for BACE1 activity, BACE1-mediated cleavage of AβPP occurs preferentially in the endosomes, which provide a suitably acidic environment [12, 19]. A previous study has indicated that BACE1 phosphorylation on Ser498 mediates its subcellular trafficking, which consequently regulates its enzymatic activity [20]. Generally, phosphorylated BACE1 is predominantly located within the TGN and the non-phosphorylated form accumulates in early endosomes [21].
SUMOylation, the attachment of an active small ubiquitin-like modifier (SUMO) to a target protein, has been implicated in many aspects of cellular pathways, including cell division, DNA damage repair, stress responses, and alteration of the subcellular localization and stability of the SUMOylated substrate [22, 23]. Multiple studies have demonstrated that SUMOylation participates in AD pathologies [24, 25]. Furthermore, BACE1 is SUMOylated on lysine residue 501 (K501), which mediates its stability and escalates its protease activity, resulting in augmentation of Aβ production [26]. A previous study has shown that feeding AD model mice a long-term high fat diet (HFD) augments Aβ aggregation [27]. However, the mechanism by which an HFD enhances Aβ accumulation and whether it mediates BACE1 post-modification is still not fully understood.
Obesity is a worldwide health problem associated with many diseases, including AD [28]. Epidemiological studies have shown that midlife obesity can increase the risk of late-life AD risk [29]. A recent study using triple transgenic AD mice indicated that a HFD enhanced neuronal oxidative stress and apoptosis by suppressing nuclear factor erythroid 2-related factor 2 signaling to augment cognitive impairment [30]. Here, we show that a 4-month HFD modified BACE1 phosphorylation and SUMOylation in APP/PS1 mice, resulting in enhanced enzyme activity and stability. Furthermore, this HFD resulted in increased Aβ plaque and cognitive impairment in these mice.
MATERIALS AND METHODS
All antibodies used in the study are listed in Table 1.
Antibodies employed in this study
pAb, polyclonal antibody; mAb, monoclonal antibody; WB, western blot; IF, immunofluorescence; IP, immunoprecipitation.
Animals
In this study, APPswe/PSEN1dE9 (APP/PS1) mice were employed after genotyping and the representative data of genotyping was shown in Supplementary Figure 1. The 2.5-month-old male APP/PS1 mice were randomly separated into two groups: 1) the normal diet (ND) group, fed a standard diet (10 kcal%fat); and 2) the HFD group, fed a high fat diet (40 kcal%fat, D12492; Research Diets). All mice were housed under standard laboratory conditions (12-h light/dark cycle) with free access to food and water. In this study, all mice were housed and maintained in the Medicine Animal Center of Jianghan University, and all experiments were carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978) and approved by the Medical Ethics Committee of Jianghan University with the approval YXLL2021-003.
Novel object recognition
After 4 months of HFD or ND feeding, mice were subjected to novel object recognition (NOR) test and Barnes maze. For NOR assay, mice were placed into an a chamber (50 cm×50 cm) with two identical objects for 5 min. The time for each mouse spent in two identical objects were recorded respectively. After 24 h, one of the two objects was replaced by a novel object with different shape and color. Mice were replaced into the same chamber to explore for 5 min again. The time for each mouse spent in novel and old object were recorded respectively. All data were recorded using the SuperMaze software (Shanghai Xinruan, China).
Barnes maze
The Barnes maze platform (91 cm diameter, elevated about 100 cm form the floor) consisted of 20 holes (each 5 cm in diameter). All holes were opened except for one target hole recognized as the recessed escape box. Spatial cues and bright light were used to motivate the mice to find the escape box. For the acquisition phase (day 1–day 4), each trail followed the same protocol with the goal to train each mouse to find the target and enter the escape box within 180 s. Mice were then allowed to remain in the box for an additional 30 s. There are two trails per day. The time spent in the escape box was recorded as the latency. In the probe test (day 5), each mouse performed one 60 s trail. The target was still located in the same position, but the escape box was taken away. The times of error to reach the target, the time of mice first find the target hole and the time spend in the target quadrant were measured and recorded using the SuperMaze software (Shanghai Xinruan, China).
Tissue collection
After behavior test, mice in the two groups were euthanized for further morphological and biochemical analyses. The mice were deeply anesthetized via intraperitoneal injection of 6%chloral hydrate (Hushi, China) and then transcardially perfused with 40 mL saline. The right hemispheres of the brains were fixed in paraformaldehyde (Biosharp, China) for the immunofluorescence assays. The left hippocampi and cortices were removed and stored at –80°C before biochemical analyses.
Tissue immunofluorescence
Brains were sliced into 30μm sections using a vibratome stage (Leica VT1000S, Germany). The brain slices were blocked with phosphate-buffered saline (PBS, Beyotime, China) containing 5%bull serum albumin and 0.1%Triton X-100 (Solarbio, China) for 1 h at room temperature. Sections were incubated overnight with different primary antibodies at 4°C, washed three times with PBS, and then conjugated with different secondary antibodies for 1 h at 37°C. After washing five times with PBS, sections were stained using Hoechst (Invitrogen, USA) for 6 min and washed three times with PBS. Finally, the slices were mounted on coverslips with an anti-fluorescence quenching agent (Beyotime, China). Images were acquired using a fluorescence microscope (Olympus, Tokyo, Japan).
Western blot
Hippocampus and cortex samples were homogenized in assay buffer (Beyotime, China) containing a protease inhibitor cocktail (Beyotime, China) and boiled for 10 min with sodium dodecyl sulfate (SDS) buffer. The Bicinchoninic acid (BCA) assay (Beyotime, China) was used for quantitative analysis of the protein homogenates. Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Millipore, USA). The membranes were blocked with 5%non-fat milk for 1 h at room temperature and then incubated overnight with primary antibodies at 4°C. Blots were detected with 1 : 3000 horseradish peroxidase-linked anti-mouse (Beyotime, China) or 1 : 3000 anti-rabbit (Beyotime, China) secondary antibodies for 1–1.5 h at room temperature. Immune bands were visualized using an enhanced chemiluminescence (ECL) system (Bio-Rad, USA) with ECL reagents (Beyotime, China), followed by quantitative analysis using ImageJ (2x, USA).
BACE1 activity assay
Briefly, fresh brain tissue was washed in cold PBS and homogenized on ice. The beta-secretase activity assay kit (Abcam, UK) was used to determine the activity of BACE1. Proteins were extracted using the beta-secretase extraction buffer. The enzyme activity was measured according to the manufacturer’s instructions. A microplate reader was used to measure fluorescence intensity. To normalize the data, the protein concentration was measured using a BCA protein assay kit (Beyotime, China).
Fluoro-Jade C (FJC) labeling
FJC labels degenerating neurons other than healthy neurons [31]. Collected right hemispheres were postfixed in 4%paraformaldehyde, cut to 30μm sections, adhered to the slides and dried for 30 min in Fragment Dryer. After completely adhered to the slide glass, the slides were immersed in an 80%alcohol solution containing 1%sodium hydroxide, followed by 2 min in 70%alcohol and 2 min in distilled water. The slides were transferred to a 0.06%potassium permanganate solution for 5 min, then to 0.0004%FJC labeling solution (Millipore, MA, USA) and washed by distilled water. The slides were cleared by xylene, dried in an oven and covered with DPX (Cat No.M100). ImageJ software (2x, USA) was used to quantitative analyze.
Transfection and cell immunofluorescence
HEK 293T cells were maintained in DMEM (Hyclone, China) with 10%(v/v) fetal bovine serum at 37°C under 5%(v/v) CO2 and 75%relative humidity. Before plasmid infection, the cells were incubated for 24 h in a 24-well plate. 500 ng of wild type BACE1 or BACE1 S498A plasmid with 0.5μL of the transfection reagent were diluted in 50μL serum free medium per well. After 48 h, the cells were collected for immunofluorescence assays.
For immunofluorescence, cells were fixed with 4%paraformaldehyde onto a slide and blocked with a phosphate buffered solution (PBS, Beyotime, China) containing 5%bull serum albumin and 0.1%Triton X-100 (Solarbio, China) for 40 min at room temperature. Slides were incubated overnight with different primary antibodies at 4°C, washed three times with PBS, and conjugated with different secondary antibodies for 1 h at 37°C. After washing five times with PBS, the slides were stained using Hoechst (Invitrogen, USA) for 6 min and washed three times with PBS. Finally, the slices were mounted on coverslips with an anti-fluorescence quenching agent (Beyotime, China). Images were acquired using a fluorescence microscope (Olympus, Tokyo, Japan).
Statistics
Data are reported as the mean±standard error of the mean (SEM). All statistical analyses were performed using Prism 7, and a two-tailed unpaired t-test. Statistical significance was set at p < 0.05.
RESULTS
HFD aggravates cognitive deficits of APP/PS1 mice
To determine the influence of a HFD on APP/PS1 transgenic mice, 2.5-month-old mice were fed a HFD until reaching 6.5 months of age. As expected, APP/PS1 mice fed with this diet for 4 months gained more perirenal fat compared to the matched ND group (Fig. 1A, B). Novel object recognition (NOR) was employed to explore the effect of an HFD on cognitive performance. No difference in preferences for two identical objects were observed between the two groups (Fig. 1C). When returned to the arena, where one of the two objects had been replaced with a novel object, the HFD-fed mice showed a significant reduction in novel object exploration than the ND-fed APP/PS1 mice, suggesting that HFD-feeding augmented memory deficits in the APP/PS1 mice (Fig. 1D). Next, we used the Barnes maze to assess spatial learning and memory. In the training phase, HFD feeding significantly decreased the learning ability, as evidenced by increased latencies to correctly identify the target over a greater number of training days compared to ND mice (Fig. 1E). In the probe test cohort, primary errors were significantly increased in the HFD-fed mice than in the ND group (Fig. 1F). Primary latencies tended to increase and time spent in the target quadrant tended to reduce in the HFD-fed mice, which indicated a reduced spatial memory compared to the ND-fed mice (Fig. 1G, H). Altogether, these results suggest that a HFD aggravated cognitive impairment in APP/PS1 mice.
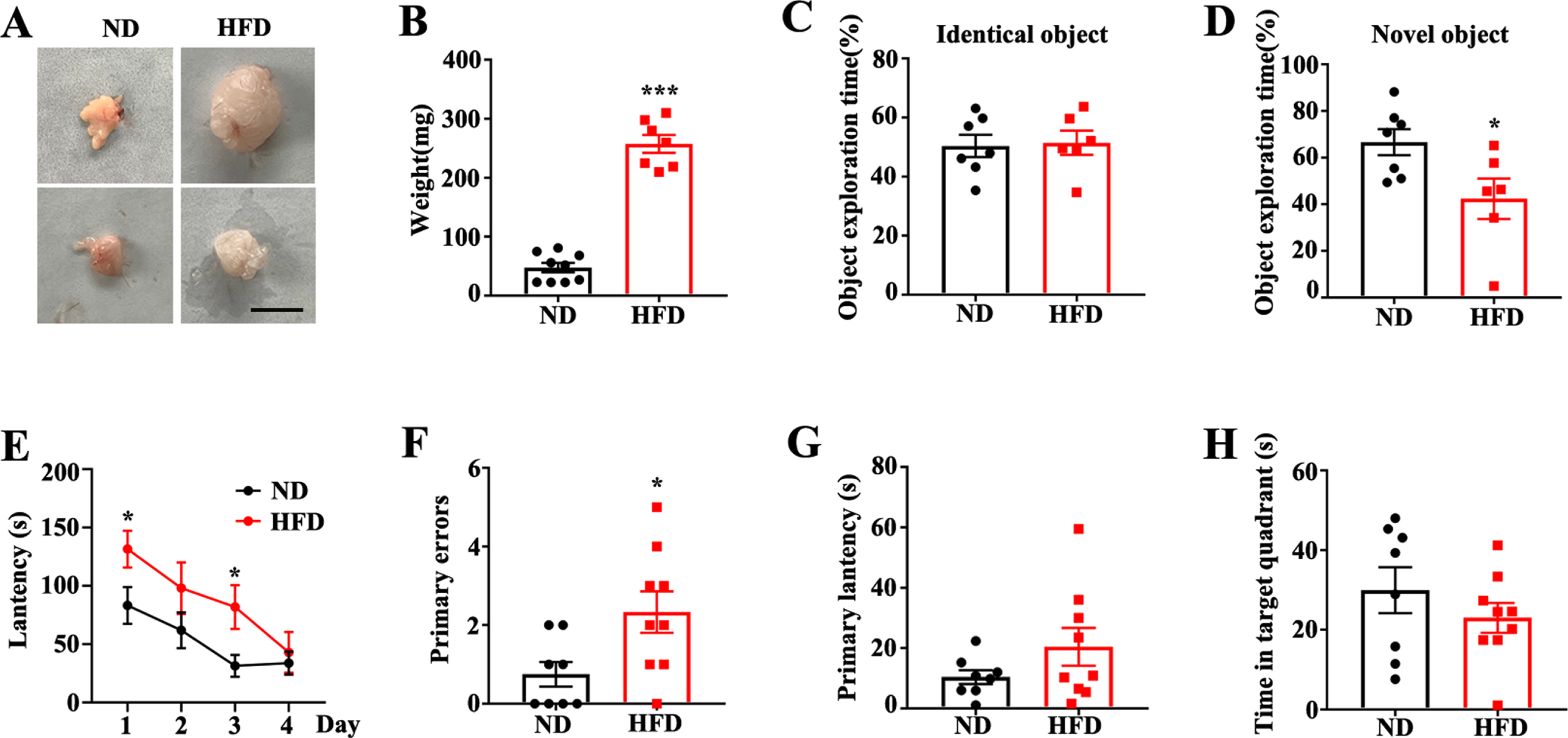
Feeding with a high fat diet aggravates cognitive deficits of APP/PS1 mice. A) Representative images of perirenal fat in APP/PS1 mice with normal diet (ND) or high fat diet (HFD) feeding (scale bar = 1 cm). B) Quantification of perirenal fat weight in APP/PS1 mice (ND/HFD n = 8/7, ***p < 0.001, two-tailed unpaired t-test). C, D) Exploration time for identical objects or new objects in novel object recognition (ND/HFD n = 7/6, *p < 0.05, two-tailed unpaired t-test). E) Exploration latency in training days of Barnes maze (ND/HFD n = 8/9, *p < 0.05, two-tailed unpaired t-test). In probe test of Barnes maze, primary errors (F), primary latency (G) and time spent in the target quadrant (H) (ND/HFD n = 8/9, *p < 0.05, two-tailed unpaired t-test). Data expressed as mean±SEM.
HFD augments impairments of dendritic integrity and neurodegeneration in APP/PS1 mice
Multiple studies have confirmed that cognitive performance relies on normal synaptic function and dendritic integrity. To further explore the potential mechanisms by which an HFD augments memory deficits in APP/PS1 mice, we evaluated the levels of dendritic integrality in the hippocampus using an immunofluorescence assay with MAP2 (dendritic marker) antibody. We found that an HFD significantly suppressed dendritic intensity in CA1 (Fig. 2A, B) and the dentate gyrus (DG) (Fig. 2A, C), although there was no effect on CA3 (Fig. 2A, D). These results highlight the adverse effects of an HFD on dendritic morphology. Normal synaptic function relies on the stable expression of synapse-associated proteins. We further evaluated synaptophysin levels using immunofluorescence. The immunofluorescent intensity of synaptophysin was significantly decreased in the DG of the HFD-fed group than that of the ND-fed group (Supplementary Figure 1C, D), whereas no significant difference was observed in the CA1 (Supplementary Figure 1A, B) and CA3 (Supplementary Figure 1E, F) regions between the two groups. Together, these results suggest that the HFD induced synaptic impairments that may result in cognitive deficits in APP/PS1 mice.
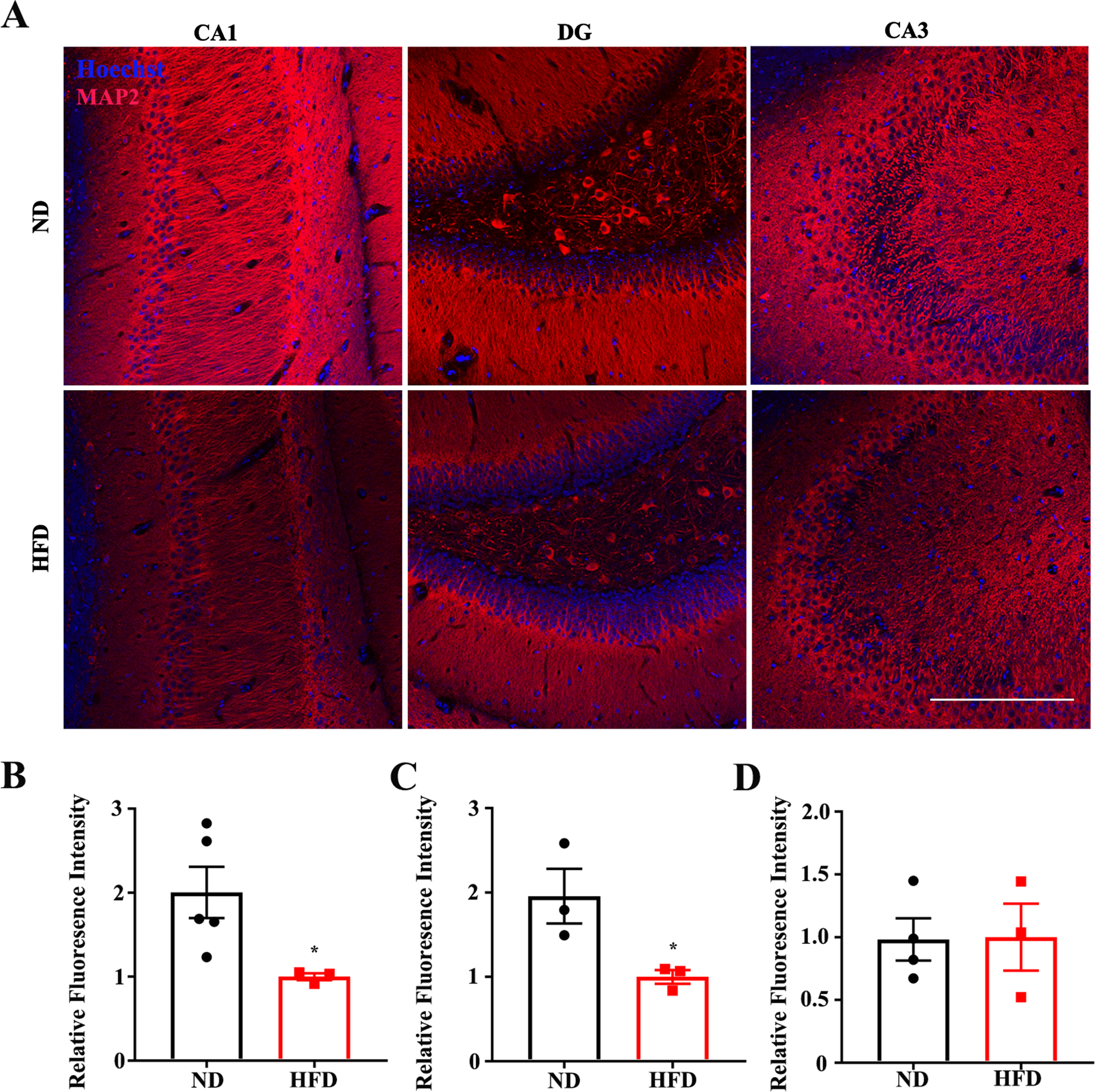
High fat diets augment impairments of dendritic integrity in APP/PS1 mice. Representative immunofluorescence images (A) and quantification of microtubule association protein-2 (MAP2) in CA1 (A), dentate gyrus (B), and CA3 (C) of normal diet (ND) or high fat diet (HFD) administrated mice (*p < 0.05, red: MAP2, blue: Hoechst, scale bar = 250μm). Data expressed as means±SEM.
A crowd of FJC-positive neurons were observed in cortical regions and in the CA1 subfields of the hippocampus in the APP/PS1 mice (Fig. 3A). As shown in Fig. 3B and C, the number of FJC-positive neuronal cells observed in HFD-fed group was significantly greater than that of ND-fed group. These results suggest HFD aggravated neurodegeneration in the AD model mice.
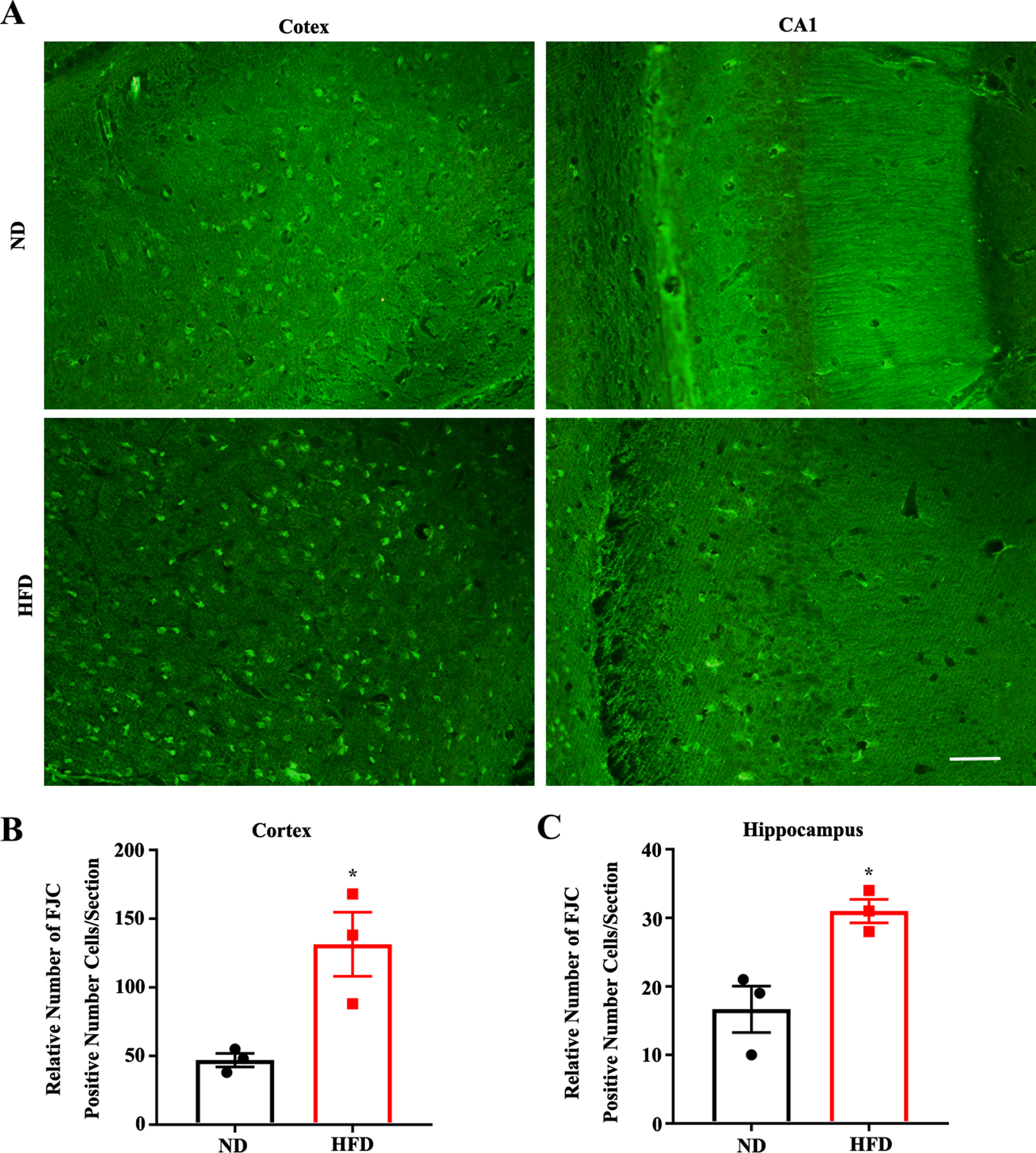
High fat diets aggravate neurodegeneration in APP/PS1 mice. Representative images of FJC staining in Cortex and CA1 of ND or HFD administrated mice (scale bar = 50μm). B, C) FJC positive cells for the groups in area Cortex and area CA1 (ND/HFD n = 3/3, *p < 0.05). Data expressed as means±SEM.
HFD enhances Aβ plaques and BACE1 activity in APP/PS1 mice
A number of studies have shown that Aβ accumulation is pivotal in the progression of AD, such as in the onset of synaptic impairment and behavioral deficits [32, 33]. Aβ plaques are observed in the brains of APP/PS1 mice at approximately 6–7 months of age [34]. To determine whether an HFD enhanced Aβ deposition, an immunofluorescence assay with anti-Aβ was used. Immunofluorescence assays and histopathological quantitative analyses indicated that both the area and number of plaques were significantly increased in the HFD-fed mice (Fig. 4A–C). As BACE1 is essential for the generation of monomeric Aβ that aggregates to form amyloid plaques, we employed assay kits to determine BACE1 activity. We found that BACE1 activity was significantly increased after HFD feeding in APP/PS1 transgenic mice (Fig. 4D). Together, these results demonstrate that an HFD is effective in increasing Aβ production and BACE1 activity in APP/PS1 mice.
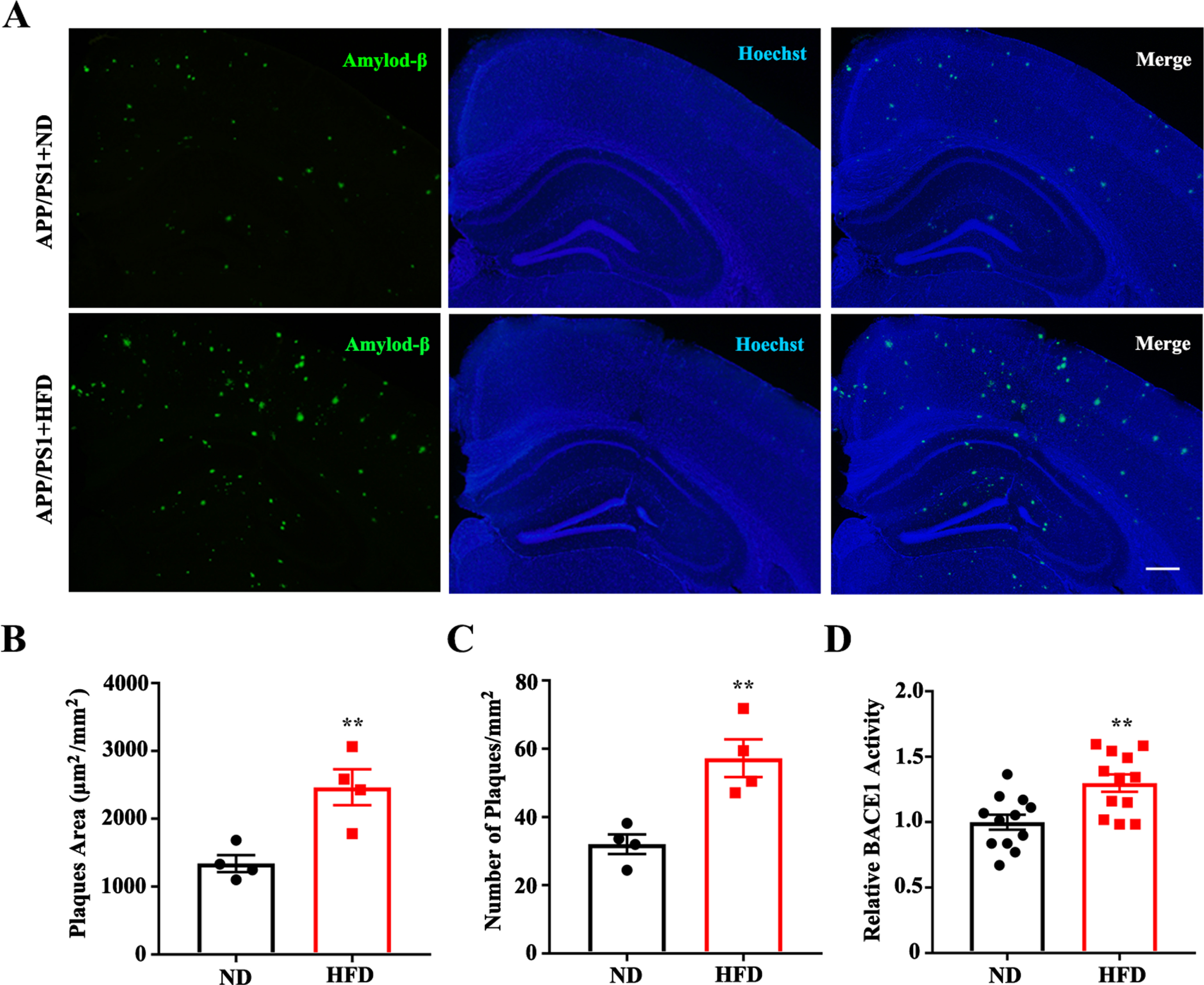
High fat diets enhance Aβ plaques and BACE1 activity in APP/PS1 mice. A) Representative images of Aβ deposits in the hippocampi and cortices of APP/PS1 mice (Green: anti-Aβ 6E10; Blue: Hoechst, scale bar = 200μm). Quantification of amyloid plaques area (B) and plaques number (C) (†ND/HFD n = 4/4, **p < 0.01). D) Quantification of BACE1 relative activity (ND/HFD n = 4/4, 3 repeats for each, **p < 0.01). Data expressed as mean±SEM.
HFD regulates BACE1 phosphorylation in APP/PS1 mice
Phosphorylation, the most extensive post-translational modification, has been proven to play an important role in regulating BACE1 activity [20, 21]. To explore the molecular mechanisms underlying the enhancement of Aβ deposits and BACE1 activity by an HFD, BACE1 phosphorylation was examined. As shown in Fig. 5A, the levels of BACE1 phosphorylation at Ser498 (BACE1 pS498) were decreased in the CA1 and CA3 regions of the hippocampus of HFD-fed APP/PS1 mice. Western blotting further confirmed that the ratio of BACE1 pS498 to BACE1 was significantly decreased in the hippocampi of HFD-fed APP/PS1 mice (Fig. 5B, C). Taken together, these results suggest that an HFD induced BACE1 dephosphorylation at Ser498, which is implicated in the regulation of BACE1 activity and Aβ production.
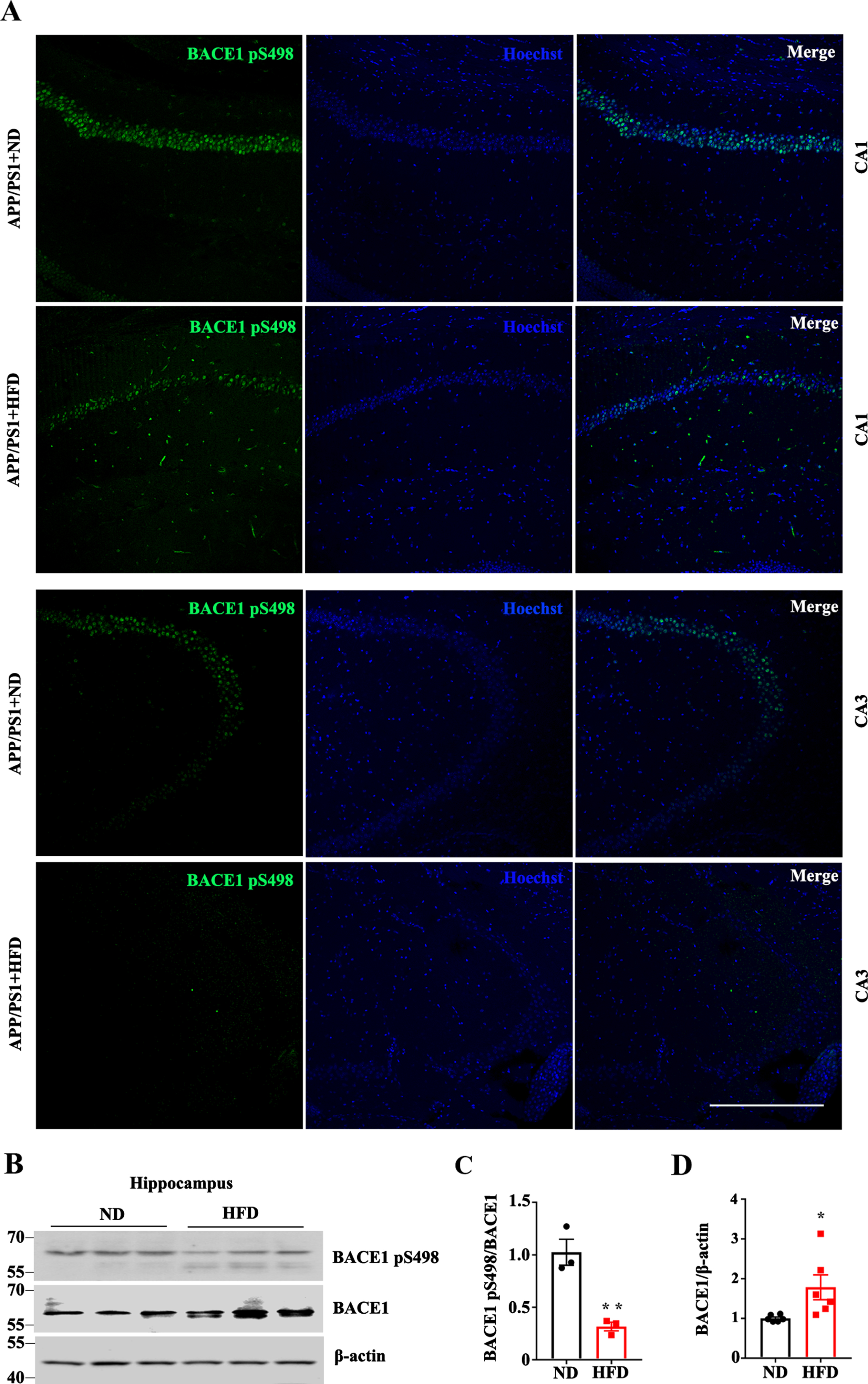
High fat diets regulate BACE1 phosphorylation in APP/PS1 mice. A) Representative immunofluorescence images with anti-BACE1 pS498 in CA1 and CA3 of ND/HFD-fed APP/PS1 mice (Green: BACE1S498P; Blue: Hoechst, scale bar = 250μm). B) Western blotting and quantitative analysis (C and D) of BACE1 pS498 and BACE1 expression in hippocampi (*p < 0.05). Data expressed as mean±SEM.
Dephosphorylation mutant regulates BACE1 trafficking between endosomes and TGN
Previous studies have demonstrated that BACE1 phosphorylation at Ser498 is involved in the regulation of intracellular trafficking, which plays a key role in the regulation of AβPP amyloidogenesis [35, 36]. To mimic BACE1 dephosphorylation induced by an HFD, we mutated BACE1 Ser498 to alanine (BACE1 Ser498A). The dephosphomimetic S498A mutant showed increased localization to the endosomes compared to the BACE1 wild type (BACE1 WT) (Fig. 6A), while also decreasing targeting to the TGN (Fig. 6B). Together, these results suggest that BACE1 dephosphorylation resulted in a defect in endosome-to-TGN retrograde trafficking and induced localization in the endosomes, where an acidic pH environment is optimal for BACE1 activity. Altogether, an HFD regulates BACE1 phosphorylation at Ser498, which further improves enzyme activity and amyloid generation.

Dephosphorylation mutant regulates BACE1 trafficking between endosomes and TGN. A, B) Compared to Flag-BACE1 WT, colocalization of Flag-BACE1 Ser498A with early endosomes increased (Red: Flag; Green: early endosomal associated protein 1 (EEA1); scale bar = 10μm). C, D) Compared to Flag-BACE1WT, colocalization of Flag-BACE1 Ser498A with Golgi decreased (Red: Flag; Green: giantin; scale bar = 10μm).
HFD modifies BACE1 SUMOylation in APP/PS1 mice
A previous study has shown that BACE1 can be SUMOylated, which would mediate its degradation and lead to augmented Aβ production [26]. Furthermore, as shown in Fig. 4B and D, the levels of BACE1 were significantly increased in the hippocampi of the HFD-fed APP/PS1 mice. To explore the underlying mechanism by which an HFD modified the expression levels of BACE1, we measured BACE1 SUMOylation levels in the hippocampus. Using an antibody against SUMO-1, we found that an HFD dramatically enhanced the bands of SUMO-1 (Fig. 7A). To confirm this, we performed immunofluorescence using an anti-SUMO-1 antibody. Compared with the ND-fed controls, significantly enhanced immunoreactivity of SUMO-1 was detected in the hippocampal CA1 region, suggesting that the presence of SUMO-1 was increased in HFD-fed mice (Fig. 7B, C). In addition, co-immunoprecipitation showed that BACE1 SUMOylation was enhanced in APP/PS1 mice after being fed with an HFD (Fig. 7D). Immunoblot assays also confirmed that an HFD increased the level of BACE1 SUMOylation, evidenced by the fact that the band at about 66 kDa was enhanced with the BACE1 antibody, which could be a ∼55 kDa BACE1 + 11 kDa SUMO band (Fig. 7E, F). Taken together, these results suggest that the HFD significantly enhanced BACE1 SUMOylation.
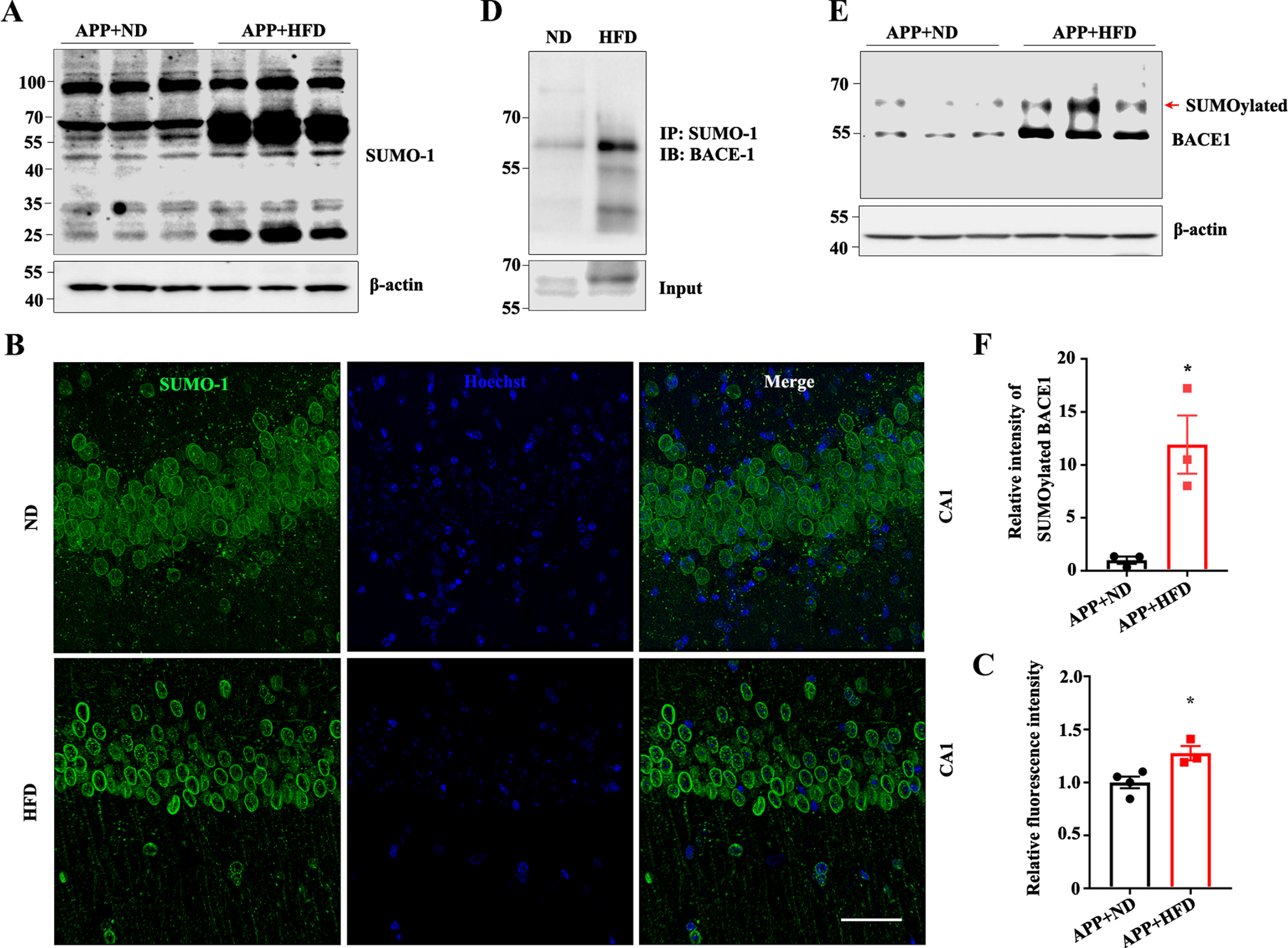
High fat diets modify BACE1 SUMOylation in APP/PS1 mice. A, B) Immunoblot of anti-small ubiquitin-related modifier-1 (SUMO-1) in the hippocampus after ND/HFD feeding. Representative immunofluorescence images (B) and quantification (C) of SUMO-1 (Green: SUMO-1; Blue: Hoechst, scale bar = 50μm, *p < 0.05). D) Coimmunoprecipitation of BACE1 SUMOylation in †ND/HFD mice. E) Immunoblot of BACE1 in the hippocampus of ND/HFD-fed APP/PS1 mice. F) Quantitative analyses of the bands around molecular weight 66 kDa for both SUMO-1 and BACE1 proteins (n = 3, *p < 0.05). Data expressed as means±SEM.
DISCUSSION
Previous clinical research has revealed that central obesity in midlife increases the risk of AD more than three decades later, independent of diabetes and cardiovascular comorbidities [37]. Aβ accumulation and deposition are critical steps in dementia progressing into AD [8]. To determine whether obesity in midlife influenced subsequent Aβ accumulation during AD pathology, 2.5-month-old APP/PS1 mice were fed an HFD for 4 months. Here, we showed that a long-term HFD modified BACE1 phosphorylation and SUMOylation, resulting in enhanced enzyme activity and stability. Additionally, an HFD impaired dendritic integrity and augmented cognitive deficits in APP/PS1 mice. These results indicated that long-term consumption of an HFD in midlife may aggravate Aβ accumulation in mice with Aβ-related mutations. There are several literatures concerning HFD leading to abnormal behavioral performance of wild-type mice [38–40]. Xu et al. showed 15-month HFD feeding exacerbated the deficits of spatial learning and memory of C57 mice at 18-month age [40]. However, the potential mechanism requires further studies.
Previous studies have demonstrated that hyperglycemia associated with obesity and a 15-month HFD causes neuroinflammation and Aβ deposition in areas of the white matter [40, 41]. Insulin-degrading enzymes in the brain, which degrade both insulin and monomeric Aβ, are reduced in the HFD-fed transgenic AD models, leading to Aβ aggregation. In addition to degradation affecting Aβ deposition, amyloidogenic processes are closely related to Aβ accumulation. Aβ peptides are derived from the successive proteolytic processing of AβPP by BACE1 and γ-secretase, and BACE1 is regarded as the rate-limiting enzyme in this process [6, 42]. To determine the effect of an HFD on amyloidogenic processes, we assessed BACE1 activity. The results indicated that BACE1 activity was significantly increased after the institution of a HFD, as shown in Fig. 4D. Thus, an HFD may induce both an increase in Aβ generation and a decrease in Aβ degradation, resulting in plaque accumulation.
Several previous studies have demonstrated that defective endosome-to-TGN retrograde trafficking widely exists in AD brains and is involved in Aβ production [20, 21]. Vps26 and Vps35, the components of the retromer complex, are significantly reduced in a brain with AD, and this blocks BACE1 retrograde endosome-to-TGN trafficking [43, 44]. Sun et al. showed that atypical protein kinase C (aPKC) inhibition led to retrograde trafficking obstruction of BACE1 [20]. Endosome-to-TGN retrograde trafficking obstruction is accompanied by enhanced levels of BACE1 in the endosome and this facilitates AβPP amyloidogenesis [20]. Phosphorylation of Ser498 is critical for BACE1 retrograde trafficking from the endosomes to the TGN, and this further influences its enzymatic activity [20, 43]. In this study, we observed that BACE1 phosphorylation at Ser498 was decreased in the CA1 and CA3 regions in the hippocampi of HFD-fed APP/PS1 mice, as shown in Fig. 5A–C. A dephosphorylation mutation of BACE1 (Ser498A) impairs its retrieval from the endosomes to the TGN and it is consequently largely retained within the endosomes. These results indicated that an HFD may enhance Aβ plaque accumulation and BACE1 activity partly via mediated BACE1 phosphorylation and subcellular localization.
BACE1 is post-translationally modified to a large extent by N-glycosylation, acetylation, phosphorylation, and S-palmitoylation [5]. Song et al. have shown that BACE1 phosphorylation of Thr252 by Cdk5 is increased, and the phosphorylation level at Thr252 is enhanced in AD brains [45]. Interestingly, phosphorylation of Ser498 by aPKC improves BACE1 retrieval from endosomes to TGN, and the reduced Ser498 phosphorylation of BACE1 contributes to AD progression [20]. Here, we showed that the phosphorylation level at Ser498 was significantly reduced in the hippocampi of HFD-fed APP/PS1 mice than in the ND-matched controls. Further investigation is needed to determine whether the changes in the phosphorylation level of BACE1 Ser498 in HFD-fed mice are regulated by aPKC.
An increasing number of studies have shown that SUMOylation is critical for normal neuronal function, and that SUMOylation imbalance is closely associated with neurodegenerative diseases [25, 47]. A previous study indicated that SUMOylation mediated BACE1 stability and increased its protease activity [26]. In the present study, we showed that BACE1 SUMOylation was significantly enhanced in the hippocampi of HFD-fed APP/PS1 mice than in the ND-matched controls. The attachment of SUMO motifs to target proteins requires a concerted series of enzymatic cascades: SUMO first binds to the E1 enzyme in an ATP-dependent manner, then transfers to UBC9 and conjugates to the consensus site of the targeted protein through E3 ligase [48]. The SUMOylation status of a specific substrate is deconjugated by SUMO protease (SENPs) [49]. Six SENPs have been identified in humans, designated as SENP1-3 and SENP5-7 [50]. Further investigation is needed to determine which SUMO-related enzymes mediate BACE1 SUMOylation in the HFD-fed mice. In conclusion, our study demonstrated that long-term feeding with an HFD mediated BACE1 dephosphorylation and increased SUMOylation, leading to aggravation of Aβ plaque accumulation and augmentation of cognitive deficits in APP/PS1 mice.