
Abstract
Background:
In the 2018 AT(N) framework, neurodegenerative (N) biomarkers plays an essential role in the research and staging of Alzheimer’s disease (AD); however, the different choice of N may result in discordances.
Objective:
We aimed to compare different potential N biomarkers.
Methods:
We examined these N biomarkers among 1,238 participants from Alzheimer’s Disease Neuroimaging Initiative (ADNI) in their 1) diagnostic utility, 2) cross-sectional and longitudinal correlations between different N biomarkers and clinical variables, and 3) the conversion risk of different N profiles.
Results:
Six neurodegenerative biomarkers changed significantly from preclinical AD, through prodromal AD to AD dementia stage, thus they were chosen as the candidate N biomarkers: hippocampal volume (HV), 18F-fluorodeoxyglucose-positron emission tomography (FDG-PET), cerebrospinal fluid (CSF), total tau (T-tau), plasma neurofilament light chain (NFL), CSF NFL, and CSF neurogranin (Ng). Results indicated that FDG-PET not only had the greatest diagnostic utility in differentiating AD from controls (area under the curve: FDG-PET, 0.922), but also had the strongest association with cognitive scores. Furthermore, FDG-PET positive group showed the fastest memory decline (hazard ratio: FDG-PET, 3.45), which was also true even in the presence of amyloid-β pathology. Moreover, we observed great discordances between three valuable N biomarkers (FDG-PET, HV, and T-tau).
Conclusion:
These results underline the importance of using FDG-PET as N in terms of cognitive decline and AD conversion, followed by HV, and could be a great complement to the AT(N) framework.
Keywords
INTRODUCTION
In 2018, the National Institute on Aging and Alz-heimer’s Association (NIA-AA) work-group published a new research framework for Alzheimer’s disease (AD), which used a scheme labeled AT(N) to further define the pathophysiology and staging of AD by characterizing research participants with various AD biomarkers using magnetic resonance imaging (MRI), amyloid positron emission tomography scan (PET), and cerebrospinal fluid (CSF) measurements [1]. This unbiased scheme plays an essential role in AD research and characterization of different disease stages [2–4]. In this AT(N) classification, A stands for biomarkers of amyloid-β deposition, T for tau neurofibrillary tangles, and N for nonspecific biomarkers of neurodegeneration or neuronal injury. Each biomarker is rated as positive (abnormal) or negative (normal) [5]. N markers are conceptualized as indicators of neurodegeneration or neuronal injury which reflect the downstream effects of AD pathology. Neurodegeneration is an important part of AD neuropathologic changes that correlate with the clinical symptoms of AD and used to stage the disease severity [6]. N markers are believed to be closely related to cognitive and behavioral manifestations of AD and provide important pathologic staging information. This current form of AT(N) framework is expandable to incorporate new biomarkers, especially N biomarkers [7]. Above all, the N biomarker group is an indispensable part of the AT(N) framework.
Nevertheless, it is still controversial which N bio-marker should be adopted. According to the recommendations, the application of three N markers [CSF total tau (T-tau), 18F-Fluorodeoxyglucose positron emission tomography (FDG-PET) hypometabolism, and hippocampus volume (HV) on MRI] were suggested, but there were differences when a different N marker was selected [1]. HV indicates cumulative loss and shrinkage of the neuropil; CSF T-tau probably reflects neuronal injury at a given point; and FDG-PET likely stands for both functional neuron impairment and loss of neuropil. Different AT(N) variants are not interchangeable. Optimal biomarker combinations for diagnosis and prediction of cognitive decline may differ by clinical stage [8, 9]. Some investigators have proposed that CSF T-tau is not a suitable candidate because it is highly correlated with CSF P-tau (Spearman’s rho > 0.90), a proposed “T” biomarker [10, 11]. The ideal N marker for AD would be reliable, reproducible, simple to measure, as well as easy to implement into large populations to better evaluate and predict the disease progression. There is also evidence suggesting that neurofilament light chain (NFL), neurogranin (Ng), and α-synuclein would likely be added to the N group [10, 13]. Our previous study suggested that progranulin (PGRN) [14, 15] and α-synuclein [16] might take part in the progression of AD, and could be candidate N biomarkers. Although an initial comparison among CSF markers of neurodegeneration including NFL, T-tau, and neurogranin has been carried out in published studies [10], currently no data regarding variable N biomarkers such as neuroimaging, CSF, and plasma biomarkers exist. Therefore, there is a need to find other potential “N” biomarkers and identify the best one.
In the present study, we aimed to 1) verify whether these biomarkers could have the potential to be candidate N biomarkers, 2) compare the selected N biomarkers by investigating their cross-sectional and longitudinal correlations with cognitive measures, and 3) the conversion risk of different N profiles, to find the best candidate biomarker for “N” in the AT(N) framework.
MATERIALS AND METHODS
Alzheimer’s Disease Neuroimaging Initiative (ADNI)
We conducted cross-sectional and longitudinal analyses of participants enrolled in the ADNI data-base (http://adni.loni.usc.edu). ADNI is a longitudinal, multicenter study launched in 2003 to assess serial changes in CSF biomarkers, blood biomarkers, neuroimaging markers, and neuropsychological assessments in three groups of elderly individuals: cognitively normal (CN), mild cognitive impairment (MCI) and AD. All AD individuals met the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria for probable AD, with Mini-Mental State Examination (MMSE) scores between 20 and 26 and Clinical Dementia Rating (CDR) global scores of either 0.5 or 1. Criteria for amnestic MCI include MMSE scores between 24 and 30, and CDR scores of at least 0.5. CN individuals had MMSE scores of 24 or higher and a CDR score of 0. All individuals were recruited from more than 50 sites across the USA and Canada. Detailed diagnostic criteria are available in http://www.adni-info.org. All the data we used were from ADNI 1, 2 and GO.
Data used in preparation of this article were obtained from the ADNI database. The study was approved by institutional review boards of all participating institutions, and written informed consent was obtained from all participants or their guardians according to the Declaration of Helsinki (consent for research).
Participants
We extracted all information from the latest merged document “ADNIMERGE.csv” updated on May 24, 2019. In our study, individuals were included if they underwent the assessments of CSF Aβ (labeled A) and CSF P-tau (labeled T). A total of 1,238 participants were recruited from the ADNI database. In further cognitive and neuroimaging analyses, 13 participants without cognitive tests and 201 without imaging data were excluded (Supplementary Figure 1).
Biomarkers of neurodegeneration or neuronal injury
Studies have examined the following potential N markers: hippocampal volume atrophy (HV), FDG-PET, CSF total tau (T-tau), plasma neurofilament light (NFL), CSF NFL, CSF α-synuclein, CSF neurogranin (Ng), CSF progranulin (PGRN), CSF soluble triggering receptor expressed on myeloid cells 2 (sTREM2), CSF Visinin-like protein 1 (VILIP-1), CSF YKL-40 (or chitinase-3-like protein 1), and synaptosome-associated protein 25 (SNAP-25) at baseline (see Supplementary Table 1) [17].
CSF measurements
In the present study, CSF Aβ42, p-tau, T-tau, and NFL were measured at the ADNI biomarker Core Laboratory (University of Pennsylvania) on the xMAP-Luminex multiplex platform (Luminex Corp, Austin, TX) using Innogenetics immunoassay kit-based reagents. CSF NFL (Unit: ng/L) was measured with a novel, sensitive sandwich ELISA method (NF-light ELISA kit, UmanDiagnostics AB, Sweden) in the University of Gothenburg, as described previously [18]. The lower limit of quantification for CSF NFL assay was 50 ng/L. Level of CSF α-synuclein was measured using LuminexMicroPlex [19]. CSF PGRN and sTREM2 (Unit: pg/mL) were measured with a MSD platform based ELISA assay, which was previously described and validated [20–22]. CSF Ng (Unit: pg/mL) was measured by electrochemiluminescence using the Ng-specific monoclonal antibody Ng7 as the coating antibody [23]. Both CSF VILIP-1and SNAP-25 were tested by a sandwich ELISA (together with the Erenna® immunoassay platform) [24]. CSF YKL-40 (Unit: ng/mL) was determined by the MicroVue YKL-40 ELISA assay at Washington University [25]. All CSF samples were performed in duplicate. Detailed information can be obtained at http://www.adni-info.org.
Plasma measurements
Blood samples were collected, centrifuged, ali-quoted, and stored at –80°C. Plasma NFL was analyzed by the single molecule array (Simoa) technique in Clinical Neurochemistry Laboratory (University of Gothenburg, Sweden) using the same methodology as previously described [26]. The plasma NFL assay used a combination of monoclonal antibodies and purified bovine plasma NFL as calibrator (details available in http://adni.loni.usc.edu). All tested samples were above the detection limit, analytical sensitivity was < 1.0 pg/mL. All samples were measured in duplicate.
Neuroimaging
Acquisition protocols and preprocessing steps for structural MRI and FDG-PET are available at http://adni.loni.ucla.edu/. Structural MRI was performed using a Vision 3.0T or 1.5T scanner (Siemens, Erlangen, Germany). Regional brain volume estimates were processed using Free-surfer software package version 4.3 and 5.1 image processing framework for the 1.5T and 3.0T MRI images, respectively. Middle temporal lobe (MidTemp) volume, entorhinal cortex thickness (Entorhinal), whole brain, ventricular volume and fusiform volume were selected for further analysis to compare the measures of brain atrophy.
FDG-PET data for each subject were pre-pro-cessed by a series of steps as described in detail elsewhere [7, 27]. In this study, the mean standardized uptake value ratio (SUVR) of previously validated AD-typical hypometabolism regions (angular, temporal, and posterior cingulate) was estimated as FDG SUVR of each participant for further analysis [27].
Cognitive scores
MMSE, Alzheimer Disease Assessment Scale 11 score (ADAS11), Alzheimer Disease Assessment Scale 13 score (ADAS13), Rey Auditory Verbal Lear-ning Test (RAVLT) Immediate, and Functional Activities Questionnaire (FAQ) were used to assess overall cognitive ability and evaluate outcome measures.
AT(N) measurements
As for A and T categories, we adopted the established cutoffs based on the ADNI database to define the diagnostic test results: positive or negative [28]. CSF amyloid positive (A+) and negative (A–) were determined by a cutoff value of 192 pg/ml for CSF Aβ42 [28]. CSF p-tau positive (T+) and negative (T–) were defined as a score above and below a cutoff value of 23 pg/ml. Binaryzation of N markers (+/–, abnormal/normal) was obtained from a Youden index-derived cutoff (ROC analyses included AD dementia as cases and CN participants as controls).
Statistical analysis
To find the best N marker(s), we conducted a three-step analysis in our study.
In the first step, we included common neurodegenerative biomarkers generated from blood test, CSF, MRI, and PET. We compared the changing trend of each N marker in the preclinical, prodromal, and dementia stages of AD: A–CN, A + CN, A + MCI, and A + AD. Then, we filtered out those non-significant marker(s) and calculated the diagnostic accuracies of selected N markers using area under the receiver operating characteristic curve (AUROC) with binary logistic regression models. Receiver operating characteristic curve (ROC) and logistic regression (LR) analyses were done using IBM SPSS Statistics 26.
Secondly, in the cross-sectional analyses, the effects of each candidate N biomarker on cognitive (MMSE, ADAS11, ADAS13, RAVLT, and FAQ) were investigated using a linear regression model. Longitudinally, the correlations of those candidate N biomarkers with cognitive performance over time were further compared by linear mixed-effects models. In the cross-sectional and longitudinal analyses, all the included biomarkers and outcome variables (cognitive scores) were all Z log-transformed to normalize the distributions, a facilitating the comparison of biomarkers. In these results, β coefficients refer to standardized effects (β= 1 implies that an increase of Z log biomarker was associated with a 1-SD increase in the dependent variable). All regression analyses were adjusted for age, gender, APOE ɛ4, years of education and diagnosis at baseline for cognitive performance.
Finally, unadjusted Kaplan-Meier (KM) analysis with the log-rank test to determine cognitive decline was performed. Clinical progression was defined as followings: 1) CN converted to MCI or AD, or their CDR scores rose to 0.5 or more, 2) MCI subjects converted to AD at follow-up or their MMSE scores decrease more than 3 points. More precisely, we conducted the subgroup analyses as follows: 1) using N markers only (N + versus N-); 2) using the combination of “A” marker and N markers, i.e., A–N–versus A–N+ versus A + N- versus A + N+. Then, we ran multivariate Cox proportional hazard models adjusted for age, gender, APOE ɛ4, and years of education at baseline.
All tests were two-tailed, and statistical significance was set at p < 0.001. All statistical analyses were performed using the R statistical software (version 3.5.1) and IBM SPSS Statistics 26.
RESULTS
Basic characteristics of the population
A total of 1,238 individuals (including 372 CN, 632 MCI, and 234 AD) were enrolled in our study. The basic demographic, clinical, and psychometric characteristics of our study population were summarized in Table 1. The total participants had a median age of 73.5 years (interquartile range IQR, 68.3, 78.1 years), a median of 16.0 years of education (IQR 14, 18 years), and a female proportion of 44.5% (Table 1). Of these participants, 782 (63.17%) were assigned to A positive (A+) group, and 644 (52.01%) were assigned to T positive (T+) group, 905 participants were categorized into AD continuum (161 A–CN, 116 + CN, A + MCI, and A + AD) when we further added the amyloid marker.
Baseline demographic characteristics of study participants
IQR, interquartile range; APOE, apolipoprotein E; A+, cerebrospinal fluid amyloid positive (CSF Aβ42 ≤ 192 pg/ml); T+, cerebrospinal fluid phosphorylated tau positive (CSF p-Tau≥23 pg/ml); A–CN, amyloid negative cognitive normal participants; A + CN, amyloid positive cognitive normal participants; A + MCI, amyloid positive mild cognitive impaired individuals; A + AD, amyloid positive Alzheimer’s disease group. *CN including SMC 95. #MCI including EMCI (Early MCI) 277 and LMCI (late MCI) 355.
Screening the candidate N biomarkers
We primarily selected several reported markers of neurodegeneration or neuronal injury: HV, FDG-PET, T-tau, plasma NFL, CSF NFL, α-synuclein, Ng, PGRN, STREM2, YKL-40, VILIP-1, and SNAP-2 (Supplementary Figure 2). We explored whether these biomarkers could be the candidate N biomarkers. N biomarkers were closely tied with aging during the preclinical, prodromal, and dementia stages of AD. We compared levels of baseline N markers from A–CN to A + CN, to A + MCI, and to A + AD (see Fig. 1a). Supplementary Figure 2 and Supplementary Table 2 showed the levels of these 12 makers in these four subgroups. To better compare their trends, combined models were showed in Fig. 1a. In this study, we found hippocampal volume (mean: A–CN 7447.31, A + CN 7317.07, A + MCI 6622.94, and A + AD 5845.55, mm3) and FDG-PET (mean: A–CN 1.33, A + CN 1.29, A + MCI 1.22, and A + AD 1.06, SUVR) declined significantly as AD progressed (p < 0.0001). Moreover, CSF T-tau, Ng, CSF NFL, and plasma NFL were also increased significantly (p < 0.0001, see Supplementary Table 2). STREM2, PGRN, α-synuclein, YKL-40, VILIP-1, and SNAP-25 did not show significant change from the preclinical to dementia stages of AD (Fig. 1a). Finally, we included six candidate N biomarkers for further analysis: N1 HV, N2 FDG-PET, N3 T-tau, N4 plasma NFL, N5 CSF NFL, and N6 Ng.
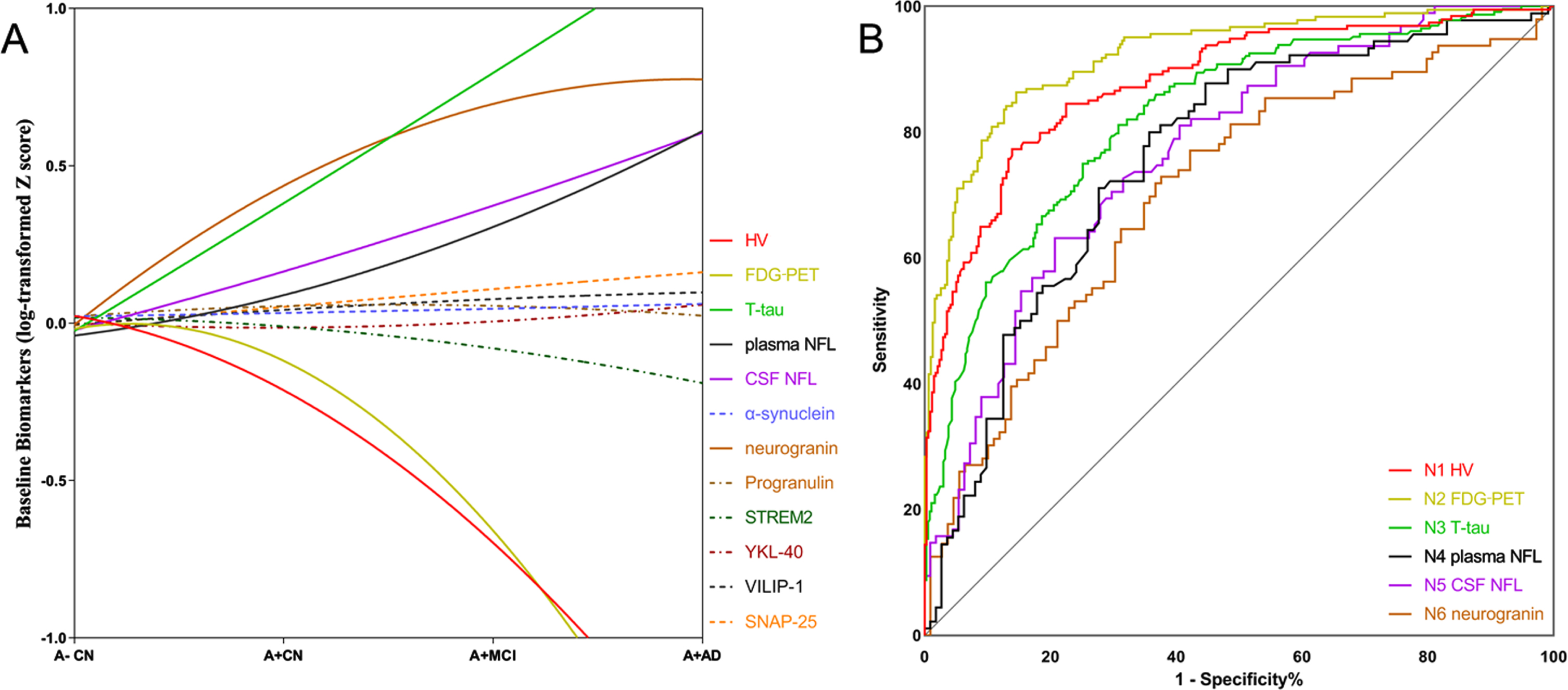
Screening the candidate N biomarkers. A) The trajectories of primarily candidate N biomarkers from the preclinical, prodromal, and dementia stages of AD. Based on the baseline levels of each biomarker (mean±SD) in four subgroups (A–CN, A + CN, A + MCI, and A + AD), we delineated an approximate trend Graph. Control: Aβ- controls (A–CN); AD continuum: Aβ+ controls (A + CN), patients with Aβ+ MCI (A = MCI), and patients with Aβ+ AD dementia (A + AD). A–indicates Aβ negative; A + indicates Aβ positive, definite A: CSF Aβ42 < 192 ng/L. B) Receiver operating characteristic curve (ROC) curves for N biomarkers for the Alzheimer’s disease (AD) cases versus cognitively normal (NC) subjects. HV, hippocampal volume; FDG-PET, 18F-fluorodeoxyglucose-positron emission tomography; T-tau, CSF total tau; plasma NFL, plasma neurofilament light chain; CSF NFL; α-synuclein, Ng, neurogranin; PGRN, progranulin; SD, standard deviation.
Accuracy of N biomarkers in predicting AD
ROC analyses of AD patients versus CN group provided cutoffs concentrations which showed the greatest diagnostic accuracy. Detailed information on the diagnostic sensitivity and specificity was summarized in Fig. 1b (Supplementary Table 3). The greatest value of the area under the ROC curve (AUC) was obtained for N2 FDG-PET (0.922). FDG-PET had the greatest sensitivity value of 86.34% and greatest specificity value of 85.44% (cutoff value, 1.199 SUVR). However, the diagnostic specificity for N1 HV was 89.09%, which was greater than the other five biomarkers (cutoff value, 6594 mm3). For T-tau, the AUC value and sensitivity value were 0.826 and 81.14, respectively (cutoff value, 74.4 pg/ml). For plasma NFL, CSF NFL, and Ng, the AUC values were 0.760, 0.768, and 0.704, respectively (see Fig. 1b). Prevalence of AT(Nx) categories based on these above N cutoffs were showed in the Supplementary Figure 1. To further compare diagnostic utilities of N markers, we compared their diagnostic accuracy in the amyloid positive subgroup. We compared those N markers in A + subgroups (A + T + and A + T-). In A + T+, the diagnostic accuracy of FDG-PET in differentiating AD from CN and MCI was much better than other markers (Supplementary Figure 3). Besides, the diagnostic accuracy of HV in differentiating AD from CN and MCI was comparable to FDG-PET. In A + T- subgroup, there were no significant differences in diagnostic accuracy between the six biomarkers (Supplementary Figure 4).
Associations between N markers and cognitive scores
In multivariable models adjusting for age, gender, years of education, APOE ɛ4, and diagnosis at baseline, the associations between cognitive scores (MMSE, ADAS11, ADAS13, RAVLT, and FAQ) and N markers were shown in Supplementary Table 4 and Fig. 2a. The levels of HV, FDG-PET, and T-tau were all correlated with all the above cognitive variables (MMSE, ADAS11, ADAS13, RAVLT, and FAQ). Notably, FDG-PET showed strongest associations with 4 cognitive variables (MMSE, ADAS11, ADAS13, and FAQ), followed by HV and T-tau (abs-olute value of β: FDG-PET > HV > T-tau > CSF NFL > plasma NFL > CSF Ng; see Fig. 2a). A few cogni-tive variables (ADAS11 and ADAS13) were associated with plasma NFL (p < 0.01) and CSF NFL (p < 0.01). No cognitive variables were associated with CSF Ng.

Associations between candidate N markers and clinical variables (cognitive scores and imaging markers). A) Association between N biomarkers and cognitive scores cross-sectionally. B) Association between N biomarkers and cognitive scores longitudinally. Beta values were all transformed to absolute values of β. All analyses were adjusted for age, gender, education, APOE ɛ4 status, and baseline diagnosis. All data were z log transformed. N1, MRI Hippocampal volume; N2, 18F-fluorodeoxyglucose-positron emission tomography; N3, CSF total tau; N4, plasma neurofilament light chain; N5, CSF NFL; N6, CSF neurogranin.
In the longitudinal analyses, the associations of all cognitive variables with HV, FDG-PET, and T-tau measures remained significant (Supplementary Table 5 and Fig. 2b). FDG-PET was closely associated with cognitive variables (abstract value of beta value: MMSE, RAVLT, and FAQ: FDG-PET > T-tau>HV>CSF NFL > plasma NFL > CSF Ng). Plasma and CSF NFL had moderate associations with these cognitive variables (p < 0.008).
Above all, three biomarkers were significantly associated with cognitive decline and neuroimaging: FDG-PET, HV, and T-tau. We also conducted a comparison between N biomarkers and brain atrophy (MRI measurements: volumes of ventricles, whole brain, entorhinal, fusiform and MidTemp), which yield similar results that FDG-PET, HV, and T-tau were best N biomarkers (see Supplementary Material). Accordingly, these three biomarkers were further compared in the following studies.
Ability of N markers to predict future clinical progression
The inter-group comparison (as evaluated with Cohen’s Kappa values) of these three valuable biomarkers (FDG-PET, MRI HV, and CSF T-tau) across diagnostic groups is shown in Fig. 3. For these three N biomarkers, there was no agreement among the diagnostic groups (all Kappa value < 0.4).
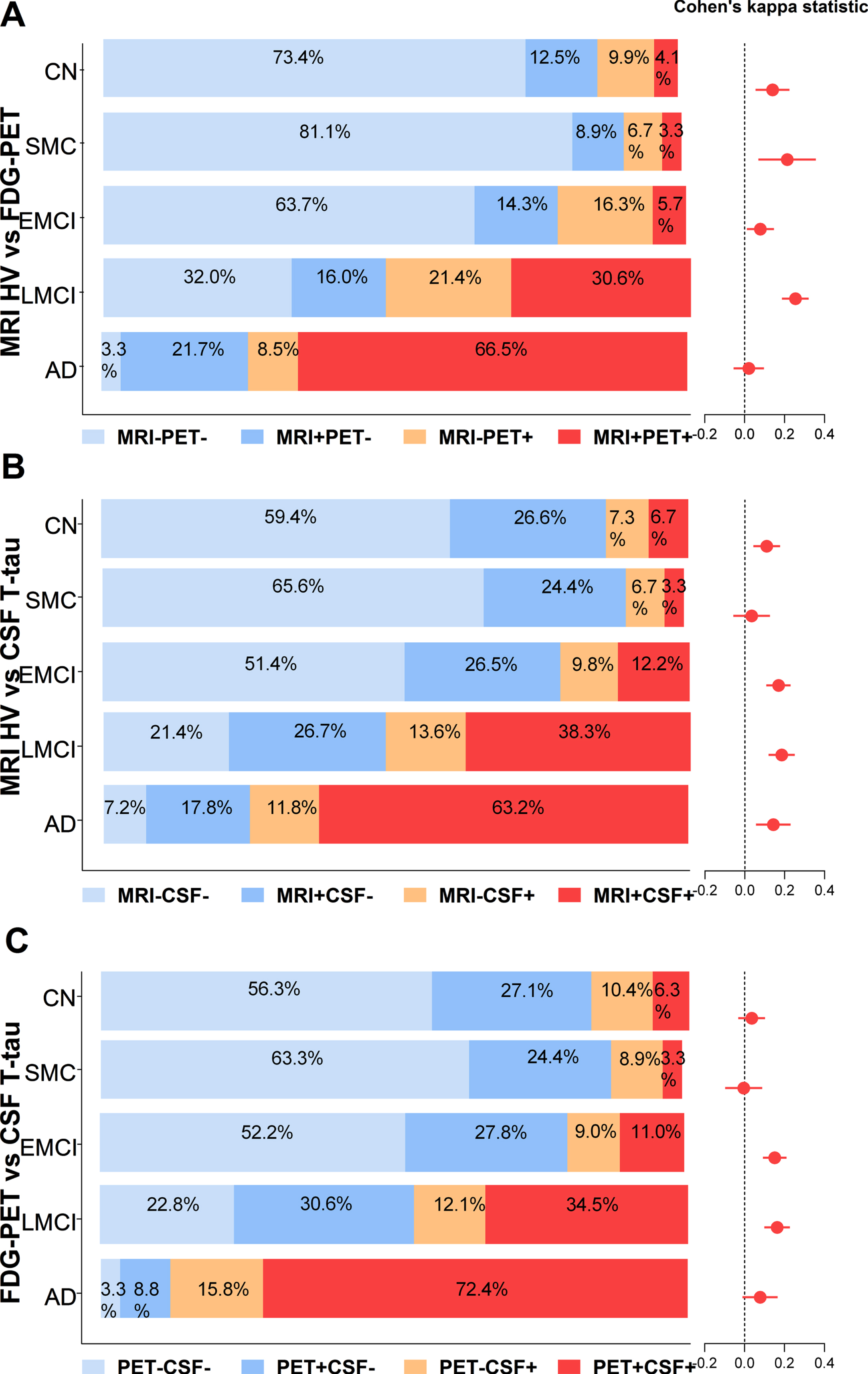
Inter-group comparison between three top neurodegeneration biomarkers in different diagnostic groups: MRI Hippocampal volume, FDG-PET, and CSF T-tau. Precentral of concordances and discordances between MRI, PET, and CSF were compared in five diagnostic groups (CN, SMC, EMCI, LMCI, and AD). A) MRI HV versus FDG-PET (concordant MRI-PET-, concordant MRI + PET + , discordant MRI + PET-, and discordant MRI-PET+). B) MRI HV versus CSF T-tau (concordant MRI-CSF-, concordant MRI + CSF+, discordant MRI + CSF-, and discordant MRI-CSF+). C) FDG-PET versus CSF T-tau (concordant PET-CSF-, concordant PET + CSF+, discordant PET + CSF-, and discordant PET-CSF+). Cohen’s Kappa statistics allowed numerical comparisons between pairs of profiles obtained using different N biomarker. Agreement was defined as coefficient values > 0.4 (fair agreement) ranging up to 1 (perfect agreement). CN, cognitively normal; SMC, subjective memory concern; MCI mild cognitive impairment; EMCI, early MCI; LMCI, late MCI; AD, Alzheimer’s disease; HV, hippocampal volume; FDG-PET, 18F-fluorodeoxyglucose-positron emission tomography; T-tau, cerebrospinal fluid total tau. MRI- indicate MRI HV negative; MRI + indicates MRI HV positive; CSF- indicate CSF T-tau negative; CSF + indicates CSF T-tau positive; PET- indicate FDG-PET negative; PET + indicates FDG-PET positive.
The results from the Kaplan-Meier analysis of N positive versus N negative and A–N–versus A–N+ versus A + N–versus A + N+ were shown in Fig. 4. Controlling for age, gender, APOE ɛ4 status, MMSE scores, and years of education at baseline, Cox proportional-hazards models were conducted to access the conversion risk. The corresponding hazard ratios were given in Supplementary Table 6. Using three N biomarkers (N1, HV; N2, FDG-PET; N3, CSF T-tau) to define N, we found that all three N + subgroups had a greater conversion rate than the corresponding N- subgroups (p < 0.0001, Fig. 4, Supplementary Table 6). The FDG-PET positive subgroup was more likely to progress than FDG-PET negative subgroup with an HR of 3.45 (95% CI = 2.50–4.77, Fig. 4a), which is greater than those of HV (HR = 2.59, 95% CI = 1.95–3.43, Fig. 4b) and T-tau (HR = 2.24, 95% CI = 1.66–3.01, Fig. 3c). When we added the A biomarker and divided participants into four subgroups (A–N–, A–N+, A + N–, and A + N+), the HRs derived for HV was bigger than FDG-PET and T-tau (p < 0.0001 HR = 3.15, 95% CI = 1.76–5.64). A + N2 + (using FDG-PET to define N2) subjects showed a 7.05-fold risk of cognitive decline compared with A–N2–individuals. We also assessed the conversion risk of cognitive impairment (Event: MMSE score decline more than 3 points) (see Supplementary Figures 6 and 7). Similarly, the results were in accordance with conversion risk of clinical progression. There was a trend that FDG-PET positive group had a faster rate of clinical progression, but when the A marker was added, HV was a better predictor for clinical progression.
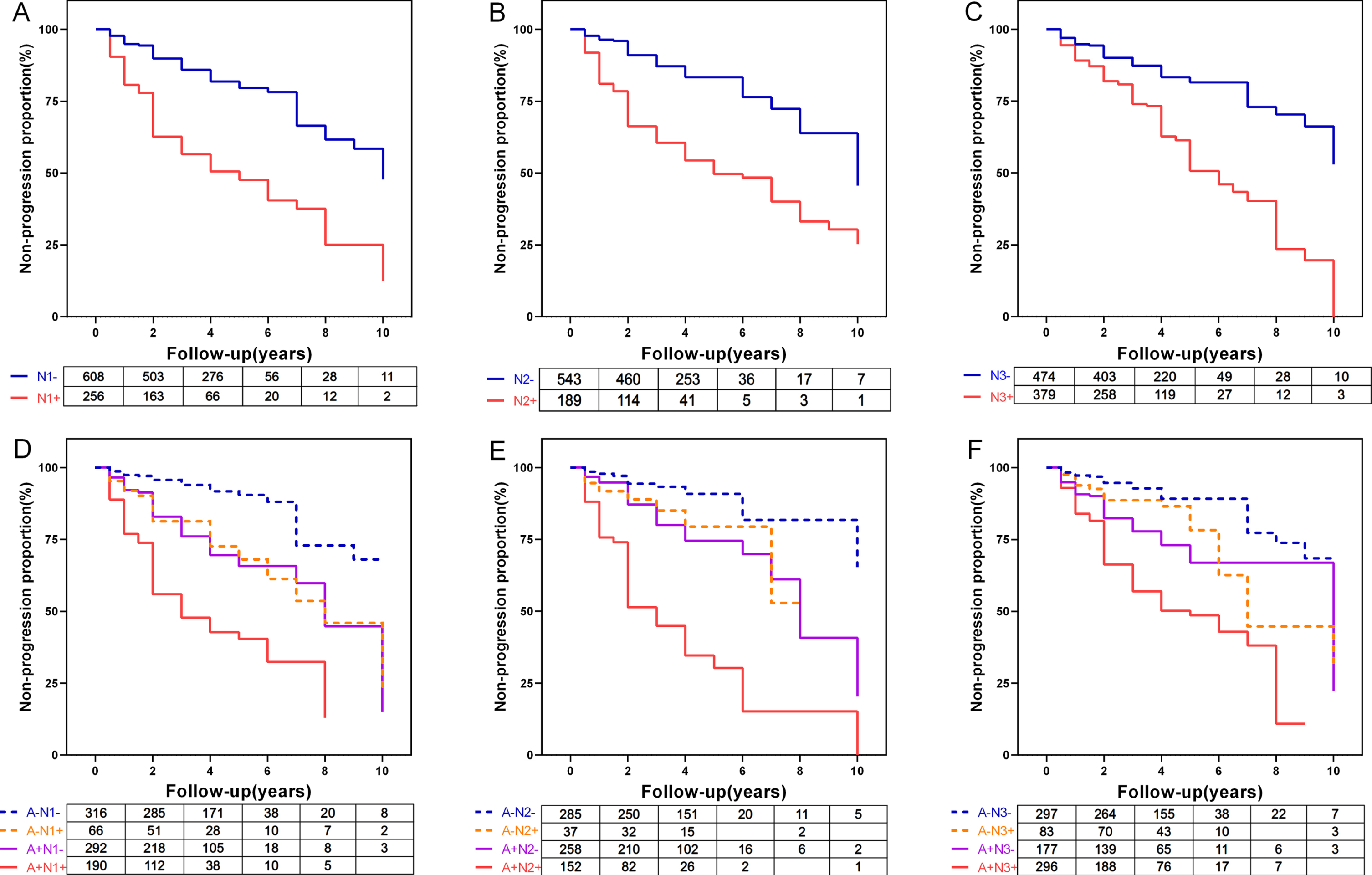
Kaplan-Meier curves showing cumulative probability of clinical disease progression. The comparisons of cumulative probability of clinical progression in (A) N1- versus N1+, (B) N2- versus N2+, (C) N3- versus N3+, and (D, E, F) add Amyloid status. The numbers of subjects at different time points were presented. N1, MRI Hippocampal volume; N2, FDG-PET; N3, CSF total tau; N-, neurodegeneration marker normal; N+, neurodegeneration marker abnormal; A–, amyloid normal; A+, amyloid abnormal.
In addition, we also compared the abilities of six N + markers (N1+, HV positive; N2+, FDG-PET positive; N3+, T-tau positive; N4+, plasma NFL positive; N5+, CSF NFL positive; N6+, Ng positive) to predict future cognitive impairment in Cox regression models (Supplementary Figure 7). No significant intergroup differences were detected among N positive subjects.
DISCUSSION
The objective of this analysis was to examine: whether there were other biomarkers could be the N markers apart from those recommended by NIAA, and which biomarker(s) could be the best N biomarker(s). In the current study, we found that 1) there were several valuable N biomarkers: HV, FDG-PET, T-tau, plasma NFL, CSF NFL, and Ng; 2) FDG-PET had greatest diagnostic utility in differentiating AD from CN (values of ROC AUC: FDG-PET > HV > T-tau > plasma NFL/CSF NFL/Ng.). Within A + T + subgroup, the diagnostic utility of FDG-PET differentiating AD and MCI from CN was still greatest; 3) HV and FDG-PET were both highly associated with cognitive declines. FDG-PET shows a closer association with cognitive decline than other markers at baseline and longitudinal analysis; 4) FDG-PET + subgroups showed more significantly cognitive decline than HV + and T-tau + subgroup. All these findings suggested that FDG-PET was a very important N marker to predict cognitive decline than other N markers, which was comparable to HV.
Neurodegenerative pathology is believed to have close associations with cognitive and behavioral manifestations of disease, act as important outcome measures in clinical trials and increase the risk of progression within a particular time frame. Our analysis evaluated the performances of N biomarkers generated from MRI, PET, CSF, and blood test. In this study, our findings supported one recommendation from NIA-AA research framework that CSF T-tau, FDG-PET, and hippocampal atrophy on MRI were proposed to be core N markers under the AT(N) scheme. Our study provided novel data-based evidence for AT(N) scheme of the new NIA-AA research framework. In the preliminary analysis, several markers (HV, FDG-PET, CSF T-tau, CSF Ng, CSF NFL, and plasma NFL) showed a significantly stepwise decrease/increase across the AD progression (A–CN, A + CN, A + MCI, and A + AD). These findings suggest that these six markers are dynamic markers that change throughout the course of AD. The inter-group comparison of three biomarkers (FDG-PET, MRI HV, and CSF T-tau) across diagnostic groups showed that these N biomarkers do not seem to be interchangeable. Our study confirmed the great interchangeability observed in other analyses between N biomarkers [29]. Our results are congruent with our expectation that there are other biomarkers with the potential to be N biomarkers, whereas the different choice of N biomarkers may result in discordances.
FDG-PET was a powerful marker of neurodegeneration in diagnosing AD, reflecting cognitive deficits and predicting clinical decline. Our results showed that FDG-PET had the greatest diagnostic utility in differentiating AD from CN than other markers even in A + T + subgroup. FDG-PET is particularly useful for early diagnosis, as it can show characteristic patterns of AD neurodegeneration earlier than MRI in individuals with MCI. Previous studies had shown the superiority of FDG-PET in early diagnosis, as it can better predict the progression of AD dementia in MCI than routine CSF or MRI tests, significantly decreasing the misclassification rate [30, 31]. Consistent with our published results, our study showed that FDG-PET was significantly associated with the severity of cognitive deficits [7]. As reported, PET allows better staging and monitoring of the extent and location of AD pathology than blood and CSF assessments [31]. These findings therefore strongly support the idea that FDG-PET can identify a wide spectrum of pathophysiological dementing conditions and visualize the distribution of neuronal injury or synaptic dysfunction. Furthermore, our longitudinal analyses discovered that the FDG-PET positive group had a faster rate of clinical progression, indicating the great value of FDG-PET in reflecting and predicting cognitive decline. This finding is consistent with a previous study suggesting that a negative FDG-PET scan strongly predicted clinical stability with high negative predictive values for both A–and A + groups [32]. FDG-PET hypometabolism, preceding MRI atrophy, is considered to be a sensitive marker of ongoing neurodegeneration dysfunction, with high accuracy in the early detection and staging of AD [31]. Cerebral hypometabolism detected by FDG-PET were reported to predict early conversion from CN to MCI and MCI to AD [33, 34]. FDG-PET performs better than SPECT and structural MRI in predicting the conversion risk from MCI to AD [34]. Another piece of evidence is that glucose hypometabolism detected by PET preceded cognitive decline and gray matter atrophy [35–37]. All these above results indicate that FDG-PET is very closely associated with the severity of cognitive deficits, making PET particularly useful for differential diagnosis, staging of disease extent, and prediction of disease progression. Thus, on the basis of our current knowledge of the advantages and disadvantages of each biomarker, FDG-PET can act as an important N biomarker in AD.
Our results also showed that HV, which was widely used to investigate structural changes in AD [38], had huge potential as an N marker for AD. Accumulating evidence indicates that HV is one of the best positioned MRI markers for clinical use [38, 39]. Hippocampal atrophy occurs in the early stage of AD, and accelerates with the progression of AD. Our findings indicated that HV had better performance in the prediction of cognitive decline than FDG-PET and T-tau. When amyloid deposition was taken into consideration in our longitudinal dataset (the biomarker “A” was added), HV is a better predictor in both A–and A + subgroups. This finding is in line with several prior MRI studies reporting that increased rates of hippocampal loss accelerated cognitive decline. HV provided important complimentary information for the prediction of cognitive decline in AD when regard of the Aβ status. Understanding discrepancies between FDG-PET and HV is essential.
CSF T-tau, one of core AD CSF biomarkers, had the strongest association with AD-related neurodegeneration than other CSF markers (CSF NFL and Ng) [10]. However, it was less robustly associated with cognition and neuroimaging outcomes when compared with HV and FDG-PET. Plasma NFL has been suggested by previous studies to be a valuable noninvasive biomarker closely related to neurodegeneration in AD patients [12, 40]. In the further analysis, we found plasma NFL was a more promising blood biomarker for neurodegeneration than some CSF biomarkers (α-synuclein, progranulin, STREM2, YKL-4, VILIP-1, and SNAP-25), but we did not find any evidence for the superiority of plasma NFL over FDG-PET, HV, or T-tau. However, larger longitudinal studies on the above-mentioned N biomarkers are still needed to further explore their advantages in predicting disease progression.
We reached an agreement on the choice of N biomarkers under the NIA–AA AT(N) research framework based on our available evidence. More recently, Mattsson et al. reported that different AT(N) variants were not interchangeable and different AT(N) combinations may influence clinical diagnosis and the prediction of cognitive decline [8]. Rather, we hope we provide a decision aid for future research and clinical decision-making when each N marker is available and different A, T and N markers can be combined in a meaningful way. We also highlight the main challenges in clinical practice and suggest research directions.
The results from this current study provide support for the proposed AT(N) scheme. A key strength of this study is that we analyzed both the cross-sectional and longitudinal associations of selected potential N biomarkers with neurodegeneration and their predictive abilities in cognitive decline in a large cohort, which facilitates the improvement of AT(N) system and the understanding of AD key pathologies. However, there were several limitations in this study. First, the sample sizes for different N biomarkers (plasma NFL, CSF NFL, and Ng) were small and significantly different, which might lead to confounding. Therefore, those findings may need to be replicated in one large-sample studies with the same sample size for different N biomarkers in the future. Second, in the present study, dichotomizing continuum markers might result in the loss of important information. Finally, although we tested comprehensive N biomarkers, we acknowledged that several other A ([18F] flutemetamol PET neocortical SUVR) and T (tau PET) biomarkers could be further tested.
In conclusion, our study suggests that levels of FDG-PET maximize the likelihood of observing and predicting significant cognitive decline over time and could be the best N biomarker. The multimodal classification of AD biomarkers (AT(N) system) is well established, but the N selection required for this approach is conflicting and there are numerous blockers to adopt this framework in clinical trials. This current study could be a complement to the AT(N) framework and have the potential effect to bridge the gap between multiple biomarker lead by AT(N) system and its clinical usage.
Footnotes
ACKNOWLEDGMENTS
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (![]() ). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
This study was supported by grants from the National Natural Science Foundation of China (91849126, 81971032 and 81801274), the National Key R&D Program of China (2018YFC1314700), ZJLab, Shanghai Center for Brain Science and Brain-Inspired Technology, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University.