
Abstract
Background:
Alzheimer’s disease (AD) is a neurodegenerative disorder and the most common cause of dementia worldwide. Despite decades of investigation, the etiology of AD is not fully understood, although emerging evidence suggest that chronic environmental and psychological stress plays a role in the mechanisms and contributes to the risk of developing AD. Thus, dissecting the impact of stress on the brain could improve our understanding of the pathological mechanisms.
Objective:
We aimed to study the effect of chronic stress on the hippocampal proteome in male APPPS1 transgenic mice and wildtype (WT) littermates.
Methods:
APPPS1 and WT mice were subjected to 4 weeks of chronic stress followed by 3 weeks of continued diurnal disruption. Hippocampal tissue was used for proteomics analysis using label-free quantitative DIA based LC-MS/MS analysis.
Results:
We identified significantly up- and downregulated proteins in both APPPS1 and WT mice exposed to chronic stress compared to the control groups. Via interaction network mapping, significant proteins could be annotated to specific pathways of mitochondrial function (oxidative phosphorylation and TCA cycle), metabolic pathways, AD pathway and synaptic functions (long term potentiation). In WT mice, chronic stress showed the highest impact on complex I of the oxidative phosphorylation pathway, while in APPPS1 mice this pathway was compromised broadly by chronic stress.
Conclusion:
Our data shows that chronic stress and amyloidosis additively contribute to mitochondrial damage in hippocampus. Although these results do not explain all effects of chronic stress in AD, they add to the scientific knowledge on the topic.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by pathological aggregation of amyloid-β, neurofibrillary tangles of hyperphosphorylated tau, neuroinflammation, cognitive impairments, and neuropsychiatric disturbances (NPDs). Despite decades of research, the etiology of AD is not fully understood, and a cure is still lacking. Until now, treatment options have been aimed at alleviating symptoms, although recently the first plaque-reducing agent (Aducanumab) was approved by the U.S. Food and Drug Administration (FDA) [1].
The development of AD is influenced by risk factors like genetic mutations, aging, and chronic or severe stress exposure [2–4]. The stress response is controlled by the hypothalamic-pituitary-adrenal (HPA) axis via the paraventricular nucleus ultimately leading to systemic glucocorticoid release (cortisol in humans and predominantly corticosterone in rodents; reviewed elsewhere [5]). Glucocorticoids have many distinct functions in the body including metabolic and inflammatory functions together with a negative feedback mechanism on the HPA axis. This negative feedback is regulated at the level of the hypothalamus, the hippocampus, and the pituitary gland, and is controlled by ligand binding to the glucocorticoid receptors (GR) [6]. The hippocampus is important for HPA axis regulation due to its high density of GRs [7]. Moreover, extensive stressful stimuli results in hippocampal volume reduction and reduced hippocampal neurogenesis [8]. Thus, the hippocampus is both a regulator and a target of stress.
AD patients present with disruption of the HPA axis evident by higher cortisol levels [9–11] which could be explained by hippocampal atrophy, and thus reduced negative feedback, or dysregulation at other levels of the axis. Higher cortisol levels in older adults with amyloid pathology correlates with faster decline on several parameters including global cognition [12]. Also, administration of glucocorticoids increased amyloid pathology both in vitro and in vivo [13].
Once activated, the GR acts as a transcription factor via glucocorticoid response element found in both nuclear DNA and mitochondrial DNA (mtDNA) [14]. Thus, mitochondrial function is directly linked to the stress response via glucocorticoid-GR mediated gene transcription. mtDNA encodes 13 mitochondrial proteins all belonging to the oxidative phosphorylation (OXPHOS) pathway responsible for the electron transport chain (complex I-IV) and ATP synthesis via the ATP synthase (complex V) and the ATP production has been coupled to GR activation [15]. Furthermore, mitochondrial function has been hypothesized to be involved in AD development with the described “mitochondrial cascade hypothesis” first presented in 2004 [16]. It hypothesizes that mitochondrial dysfunction is a key driver of AD development and progression, which may be due to potential compromised inherited mtDNA and the accumulating environmental “hits” on mtDNA throughout the lifespan (reviewed elsewhere [17, 18]), which would make the mitochondria a target in AD drug development [19–22]. Altogether, it appears that mitochondrial function, environmental stress exposure and AD pathology are intertwined, although the exact mechanisms are only partly understood.
Over the last decades, mass spectrometry-based proteomics approaches have proven to be a powerful strategy in basic research, biomarker discovery and drug hunting for instance addressing neuroinflammatory alterations under different pathological conditions [23]. We have chosen to leverage this expertise on our study.
The aim of this study was to use quantitative bottom-up proteomics to examine changes in the hippocampal proteome of aged APPPS1 transgenic mice and wildtype (WT) littermates subjected to chronic stress. We focused on the hippocampus because of its vulnerability to stress, high involvement in HPA axis regulation and its role in cognition and emotional regulation of mnemonic processes as well as AD development. We found that chronic stress induced dysregulation of proteins annotated to the OXPHOS, AD and metabolic pathways. Especially, complex I of the electron transport chain was compromised in WT mice exposed to chronic stress while APPPS1 mice exposed to chronic stress showed more widespread dysregulations of the OXPHOS pathway. Lastly, we put forward a hypothesis on the relationship between chronic stress, mitochondrial function, neuroinflammation and behavioral outcome in AD.
MATERIALS AND METHODS
Transgenic animal model
The APPPS1 mouse model expresses the human APP mutations (Swedish K670M and N671L) and the human PSEN1 mutation (L166P) on a C57BL/6J background strain [24]. All animals were purchased from Charles River, Germany. For this study, we used 6-month-old male APPPS1 and wildtype (WT) littermates. All animals were housed individually upon arrival and received food and water ad libitum except for during the deprivation periods in the chronic stress paradigm. Room humidity was 55% ±5% and room temperature was 21±2°C. Lighting conditions were controlled to 12L/12D with lights on at 6.00 am, except for the groups subjected to the chronic stress paradigm, which were kept under diurnal disruption conditions (see Chronic stress paradigm). All in vivo experiments were approved by the veterinarian team and in accordance with European Communities Council Directive no. 86/609, the directives of the Danish National Committee on Animal Research Ethics, and Danish legislation on experimental animals (license no. 2014-15-0201-00339 C01 and C05).
Nest building activity
Prior to the chronic stress paradigm, nest building activity was evaluated to demonstrate a phenotypic difference between WT and APPPS1 mice and to ensure that mice performed according to previous observations. This assay was performed as previously described [25, 26]. Briefly, on the test day standard nesting material was replaced by a rectangular cotton nestlet 1 h into the light phase (7.00 am). Nesting activity was scored hourly for 8 h and again 24 h after initiation and based on the degree of shredding and coverage of igloo openings. The score range was from 0 (no shredding and no coverage) to 8 (100% shredding and coverage of all 3 igloo openings).
Chronic stress paradigm
Mice were randomly divided into either chronic stress (Stress) or control (Ctrl) groups (n = 10/group) after the nesting activity assay. Stress groups were subjected to 28 days of chronic stress using one stressor a day (food deprivation, water deprivation, cage tilt, or confinement to small container) in combination with chronic diurnal stress with 10 h light and 10 h darkness (10L/10D, first day lights on at 6.00 am) as described previously [25]. To examine chronic changes of the stress exposure, we euthanized all animals 3 weeks after end of the chronic stress paradigm. During this time the stress groups were kept on the 10L/10D lighting (see Fig. 1A for schematic illustration of experimental workflow).

A) Schematic illustration of experimental in vivo setup and ex vivo workflow. Animals were subjected to the nest building assay (NEST) prior to the random assignment to either chronic stress (Stress) or control (Ctrl) groups. Animals were then subjected to 4 weeks of chronic stress followed by 3 weeks of 10L/10D lighting before euthanization. Left hippocampi were isolated and processed for liquid chromatography with tandem mass spectrometry, followed by statistical data analysis and bioinformatic analysis. Plasma was collected for corticosterone levels and right hemispheres were used for quantification of plaque pathology via Thioflavin S staining. B) Plasma corticosterone levels were determined, and two-way ANOVA analysis revealed no difference between groups (n = 10/group). C) Nest building activity revealed a significant main effect of genotype ($$p < 0.0013) and time (# # # #p < 0.0001) together with a significant interaction effect (∘ ∘ ∘ ∘p < 0.0001). Statistical analysis was performed using two-way ANOVA with repeated measures and n = 20/group.
Euthanization and tissue harvesting
Mice were brought to the procedure room immediately before euthanization by decapitation as described previously [25]. All mice were euthanized in the middle of the light phase. Plasma was collected for corticosterone determination and brains were quickly removed and hippocampi dissected on ice and snap frozen on dry ice. All samples were stored at –80°C until further use. In the spirit of NC3R [27], excess accrued hippocampal tissue, from a larger study designed for a different experimental question, was used to generate the data reported here.
Sample preparation
Hippocampal tissue samples (n = 10/group) were homogenized in 5% sodium deoxycholate (SDC) using 0.4–1.2 stainless steel beads and Bullet Blender GOLD (Next Advanced, USA) for 10 min at maximum speed at 4°C. Protein concentrations were determined using BCA protein assay (ThermoFisher Scientific, USA) according to manufacturer’s instructions.
Hippocampal homogenates were prepared as previously described [28] with slight alterations. Briefly, 100μg homogenate was placed in filter-aided sample preparation (FASP) YM-10 kDa spin filters (Millipore, Billerica, MA, USA) with 0.5% SDC and centrifuged at 14,000 g for 15 min. Filters containing proteins were then incubated for 30 min at 37°C in digestion buffer (0.5% SDC, 50 mM TEAB (triethyl ammonium bicarbonate), 10 mM TCEP (tris(2-carboxylethyl)phosphine), 50 mM CAA (chloroacetamide)). After an additional centrifugation, filters were washed with digestion buffer and centrifuged again. Samples were then digested using 2μg of trypsin dissolved in digestion buffer and incubation at 37°C overnight. Digested proteins were eluted in 50 mM TEAB. Remaining SDC was removed from samples by phase separation using two washing steps with ethylacetate containing 1% trifluoroacid and centrifugation at 14,000 g for 1 min. Lastly, samples were dried by vacuum centrifugation and stored at –80°C until use.
LC-MS/MS
Hippocampal samples were analyzed using an automated liquid chromatography-electrospray ionization MS/MS system with an ultraperformance liquid chromatography system (Dionex RSLCnano Proflow, Amsterdam, The Netherlands). The system is coupled to an emitter for nanospray ionization (NewObjective, USA) into a quadrupole Orbitrap mass spectrometer (ThermoSci Q Exactive HF-X, Thermo Scientific, Waltham, MA, USA). 2μg of each trypsinized sample were loaded onto a 96-well loading plate in random order and loaded automatically on a reversed C18 phase column (Dionex; Acclaim PepMap100 C18, 5μm precolumn and 75 cm Acclaim Pepmap RSLC, 75μm ID main column (Thermo Scientific)). Samples were eluted with a linear gradient of 96% solvent A (1% FA) and 4% solvent B (acetonitrile) increasing solvent B to 35% on a 60-min ramp gradient.
Corticosterone ELISA
To determine plasma corticosterone levels, we used the commercially available Corticosterone Enzyme-Linked Immunosorbant Assay (ELISA) Kit (Catalog number K014-H5, Arbor Assays Inc, USA) and followed manufacturer’s instructions.
Thioflavin S staining
Cryostat sectioning and Thioflavin S staining of right brain hemispheres were performed as previously described [25, 29]. Briefly, coronal sections (20μm) of prefrontal cortex and hippocampus were mounted to SuperFrost plus glass (VWR, Denmark; 5 sections/glass). Sections were dehydrated and stained with 1% aqueous Thioflavin S (T-1892, Sigma Aldrich, Denmark). Images were acquired using Leica DM5500 B upright microscope equipped with Leica DFC450 camera using 2.5 X object lens (Leica, Denmark), while quantification of plaque number and load were performed using ImageJ.
Data processing and statistics
MaxQuant version 1.6.8.0 [30] was used to process raw data files. The FASTA file contained the UniProt mus musculus database (AUP000000589) and was downloaded on October 3, 2019. The database was modified to include protein sequences of the human APP and PSEN1 with the appropriate mutations due to the transgenic APPPS1 mouse model used in this study. Statistical analyses were performed by Biogenity ApS, Denmark, using R, and was only performed for proteins with 50% or more valid values in at least one sample group. 10 samples per group were used for this study and significance level was set at p < 0.05. Comparisons were made between APPPS1 Ctrl versus WT Ctrl, APPPS1 Stress versus WT Ctrl, WT Stress versus WT Ctrl, and APPPS1 Stress versus APPPS1 Ctrl. Functional Enrichment Analysis and Protein-Protein Interaction Network analysis were performed using STRING (July-September 2021) [31, 32]. Statistical analyses and graph illustrations of nesting activity, thioflavin S and corticosterone data were performed using GraphPad Prism v.9 and two-way ANOVA with repeated measures, unpaired t-test and two-way ANOVA, respectively.
RESULTS
Transgene-induced nesting deficits
Prior to the chronic stress paradigm, general well-being of APPPS1 and WT mice were evaluated using the nesting activity assay. We found a significant main effect of genotype (F1,38 = 12.15, p = 0.0013) and time (F8,304 = 48.59, p < 0.0001), together with a significant interaction effect (F8,304 = 12.65, p < 0.0001). This suggests that APPPS1 mice are significantly impaired in their nesting behavior as we have previously shown [25]. All APPPS1 mice illustrated this robust deficit compared to WT mice, and thus no APPPS1 mice were excluded from further experiments. Nesting deficits may be linked to damage to the hippocampus, as shown in hippocampal lesion models [33], or AD-like pathology [34].
Stress-induced plaque pathology
Chronic stress is considered a risk factor in AD and has been shown to increased AD-like pathology in vitro and in vivo [11, 35–37]. Using Thioflavin S staining of prefrontal cortex and hippocampus from the APPPS1 cohort, we found that chronic stress exposure increased plaque pathology in prefrontal cortex non-significantly (p = 0.0535 plaque load and p = 0.1130 plaque count, Fig. 2B, D). Also, we found no change in plaque load or plaque count in hippocampus after exposure to chronic stress (Fig. 2A, C).
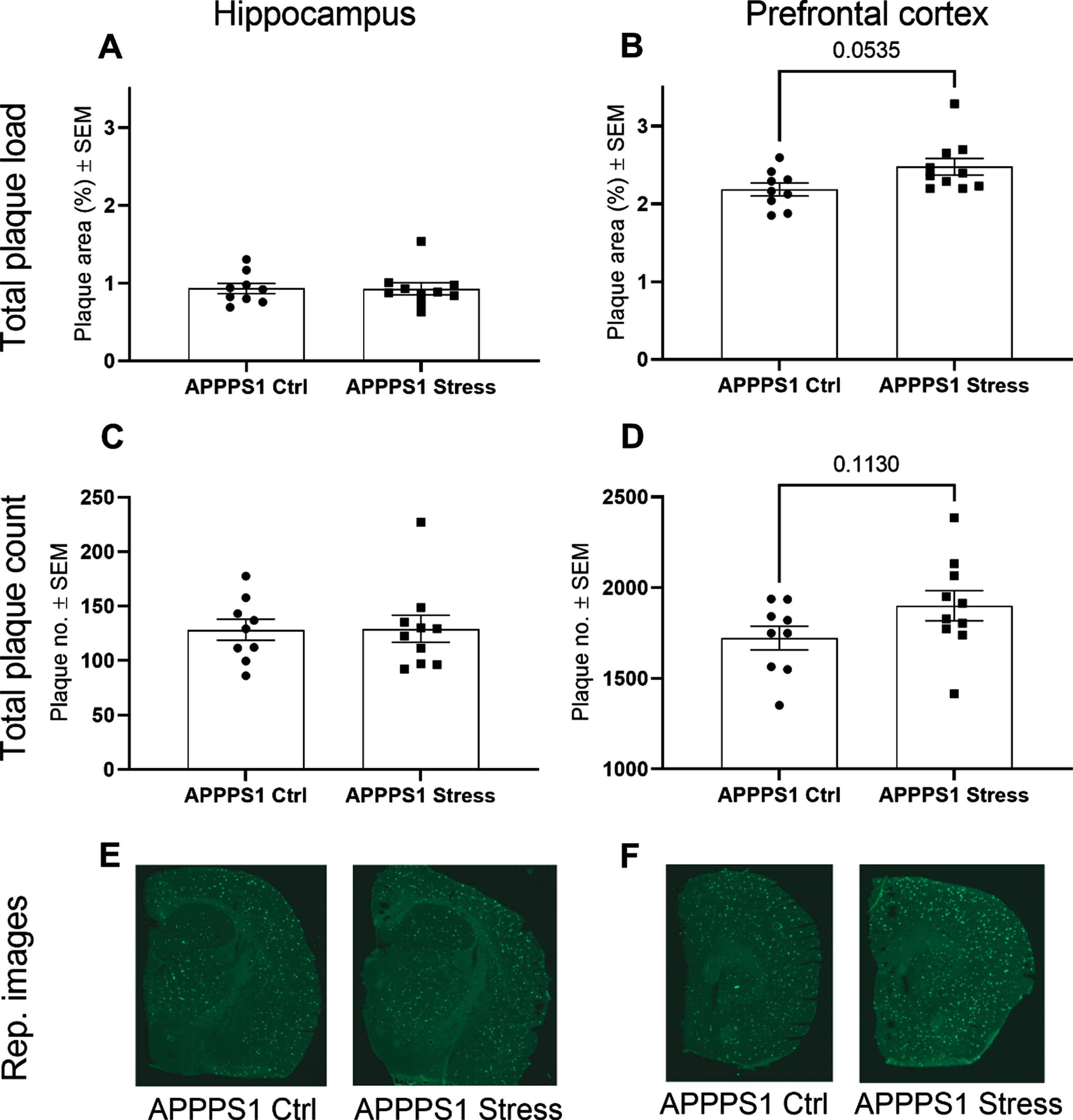
Impact of chronic stress exposure on plaque pathology in prefrontal cortex and hippocampus. Thioflavin S staining revealed a trend of increased plaque pathology in prefrontal cortex following chronic stress exposure (p = 0.0535 (B) and p = 0.1130 (D)) but no change in plaque pathology in hippocampus (A, C). Representative images of Thioflavin S staining of PFC (E) and hippocampus (F). Statistical analyses were performed using Mann-Whitney test except for analysis of prefrontal plaque count, which was analyzed using unpaired t-test (n = 9-10/group).
Plasma corticosterone
In the current study, we found no statistically significant effect of chronic stress on basal plasma corticosterone levels measured 3 weeks after the end of the chronic stress protocol (Fig. 1B), which is in line with our previous observations [25].
Unique identifications
The hippocampal tissue was subjected to bottom-up quantitative analysis. After protein identification at 1% false discovery rate (FDR) and stringent systematic filtration for missing values, p-value (< 0.05) and > 2 unique spectral matches (PSMs), we identified 1,153 quantifiable unique proteins. Of these, 1,106 were identified in all groups while the remaining 47 proteins were not found in at least one group (Fig. 3D and Supplementary Table 1). Three proteins were unique to WT Ctrl (MRS2, UGGT1, and FXYD6). 7 proteins were found only in Stress groups (1 in WT Stress (AGL), 3 in APPPS1 Stress (PPP3CB, NDUFB7, ARL3), and 3 in both APPPS1 Stress and WT Stress (FARP1, GAPVD1, MYH9)). Two proteins were found only in the APPPS1 Ctrl group (S100A6, AFG3L2).
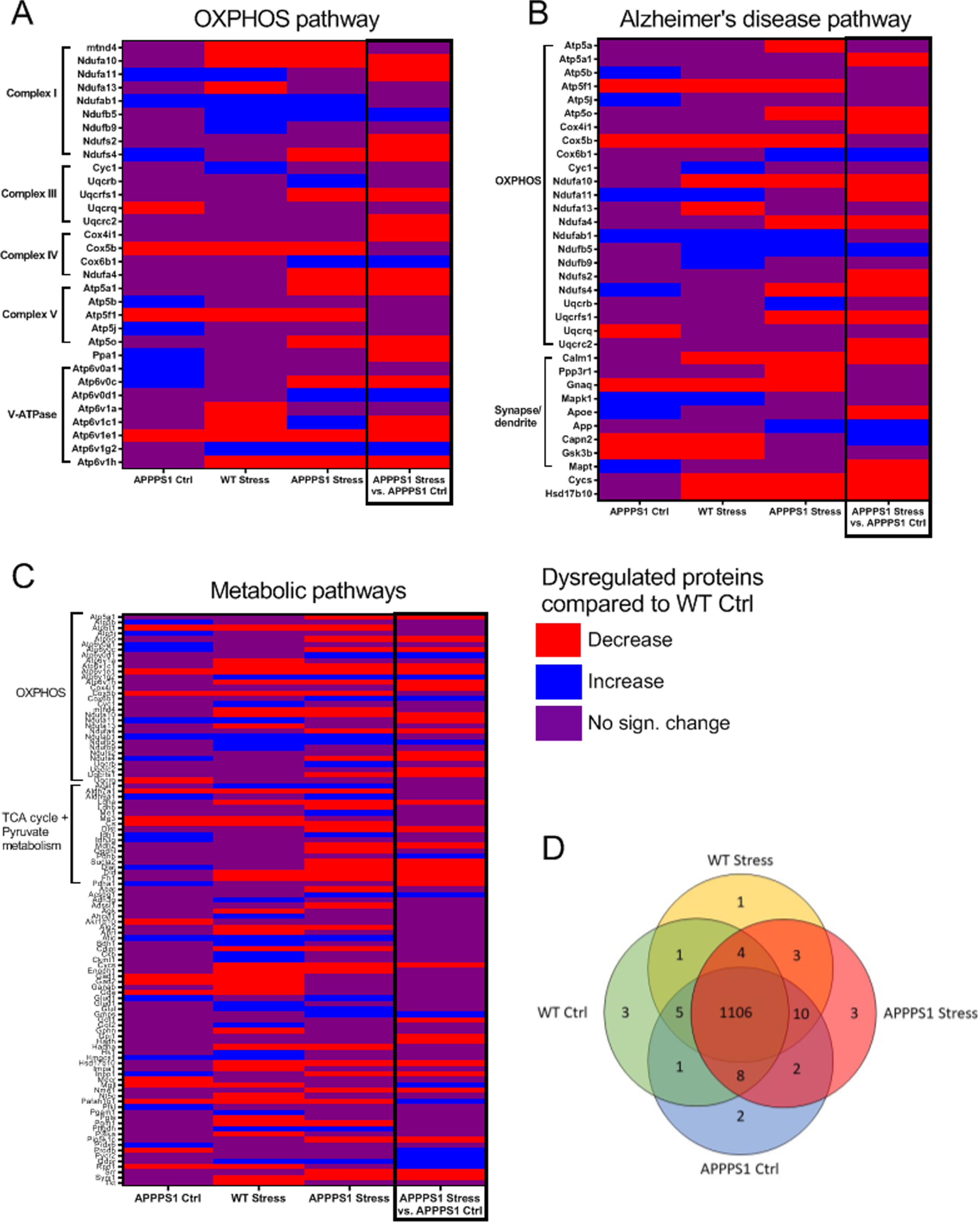
Graphic illustration of significantly dysregulated identifications compared to WT Ctrl (or APPPS1 Ctrl) and 3 identified KEGG pathways. A) OXPHOS pathway with proteins annotated to the specific complexes of the pathway. B) Alzheimer’s disease pathway with proteins annotated to the OXPHOS pathway and synapse/dendrite cellular component. C) Metabolic pathways with proteins annotated to the OXPHOS pathway and TCA cycle + pyruvate metabolism. D) Venn diagram of proteins uniquely found in specific groups. Identified pathways were found using STRING Functional Enrichment Analysis. All comparisons were between respective groups and WT Ctrl except for APPPS1 Stress versus APPPS1 Ctrl to the far right (black box). n = 10/group.
Between APPPS1 Stress and APPPS1 Ctrl, we found 198 significantly regulated proteins (79 upregulated and 119 downregulated). 82 proteins were significantly upregulated in WT Stress compared to WT Ctrl and 211 proteins were downregulated. We observed 61 significantly upregulated proteins in APPPS1 Ctrl compared to WT Ctrl and 117 downregulated. Lastly, 84 proteins were significantly upregulated in APPPS1 Stress compared to WT Ctrl, while 177 proteins were significantly downregulated (see Supplementary Tables 2–5 for specifications).
Pathway enrichment analyses
We further investigated the relationship between the significantly up- and downregulated proteins using available STRING v11.0 functional enrichment analyses that uses literature based Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis [31, 32]. We identified 16 KEGG pathways among the significantly upregulated proteins in APPPS1 Stress compared to APPPS1 Ctrl and 40 pathways among the downregulated proteins. Based on FDR values, the top 10 KEGG pathways are presented in Table 1 for both up- and downregulated pathways. The analysis of WT Stress compared to WT Ctrl identified 94 KEGG pathways of upregulated proteins and 79 KEGG pathways of downregulated proteins. Top 10 pathways of up- and downregulated proteins are presented in Table 2. Between APPPS1 Ctrl and WT Ctrl, we identified 71 KEGG pathways of upregulated proteins and 29 KEGG pathways of downregulated proteins, of which the top 10 pathways are presented in Supplementary Table 6. Lastly, between APPPS1 Stress and WT Ctrl, we identified 26 KEGG pathways of upregulated proteins and 48 KEGG pathways of downregulated proteins. Supplementary Table 7 represents the top 10 KEGG pathways of up- and downregulated proteins.
Top 10 KEGG pathways of significantly upregulated and downregulated proteins in APPPS1 Stress compared to APPPS1 Ctrl. Analysis performed by STRING Functional Enrichment Analysis
Top 10 KEGG pathways of significantly upregulated and downregulated proteins in WT Stress compared to WT Ctrl. Analysis performed by STRING Functional Enrichment Analysis
Proteins annotated to oxidative phosphorylation (OXPHOS) were found in the top 10 STRING-identified KEGG pathways of all comparisons except the downregulated protein comparison between APPPS1 Ctrl and WT Ctrl. Closer examination into this pathway identified proteins belonging to four of the five complexes of the respiratory chain and to the vacuolar-type ATPase (v-ATPase; Fig. 3A). The V-ATPase pumps protons into the lumen of organelles like vesicles, lysosome and endosome via ATP hydrolysis thus producing an acidic intraluminal environment. The KEGG pathway Alzheimer’s Disease were likewise identified in all comparisons (Fig. 3B). Of the proteins annotated to this pathway, 23 were also annotated to OXPHOS while 9 proteins (CALM1, PPP3R1, GNAQ, MAPK1, APOE, APP, CAPN2, GSK3B, MAPT) were annotated to the cellular compartment Synapse/dendrite (Gene ontology). Lastly, metabolic pathways were also among the top 10 KEGG pathways in all comparisons (Fig. 3C) and here 31 proteins were also annotated to OXPHOS and 19 proteins were annotated to the TCA cycle and pyruvate metabolism.
Compared to WT Ctrl, APPPS1 Ctrl mice had 12 dysregulated proteins annotated to OXPHOS (ATP6V0C, PPA1, ATP6V0A1, ATP5J, ATP5B, NDUFA11, NDUFAB1, NDUFS4, UQCRQ, COX5b, ATP5F1, ATP6V1E1), WT Stress had 15 dysregulated proteins annotated to this pathway (ATP6V1E1, ATP6V1C1, ATP6V1A, ATP6V1H, ATP5F1, NDUFA13, NDUFA10, MT-ND4, COX5B, ATP6V1G2, CYC1, NDUFA11, NDUFAB1, NDUFB5, NDUFB9) while APPPS1 Stress had 19 dysregulated proteins annotated to this pathway (ATP5A1, ATP5FL, ATP5O, ATP6V0C, ATP6V0D1, ATP6V1C1, ATP6V1E1, ATP6V1G2, ATP6V1H, COX5B, COX6B1, MT-ND4, NDUFA10, NDUFA4, NDUFAB1, NDUFB5, NDUFS4, UQCRB, UQCRFS1). Interestingly, in the WT Stress group most of these proteins belonged to Complex I of the respiratory chain and the V1 domain of the V-ATPase complex, while dysregulated proteins of the APPPS1 Ctrl and APPPS1 Stress groups were more widespread across the OXPHOS pathway. Therefore, it is likely that chronic stress exposure more directly impacts complex I and the V1 domain of the V-ATPase complex in the non-amylogenic brain, as previously shown by [38], while amyloid pathology impacts the OXPHOS pathway more widely, as previously shown by [39, 40]. Altogether, our data analysis suggests that both amyloid pathology and chronic stress exposure may contribute to dysregulation of OXPHOS and that both factors together have an additive effect on this dysregulation.
DISCUSSION
We observed that chronic stress exposure had significant impact on the mitochondrial function in both WT and APPPS1 mice. In particular, the energy producing pathways like OXPHOS, TCA cycle, and pyruvate metabolism were highlighted in the bioinformatic analyses. Reduced respiratory activity of mitochondria after exposure to chronic mild stress was reported in mice hippocampus, hypothalamus, and cortex [41] and synaptic mitochondrial dysfunction, including the OXPHOS pathway, was likewise linked to chronic mild stress exposure in rat hippocampus [42]. Similar findings have been reported for prefrontal cortex (PFC) and nucleus accumbens in mice following a multimodal chronic restraint stress paradigm [43]. Furthermore, this study showed that the dysregulated proteins encoded by the mtDNA were all upregulated in the PFC of stressed mice and major depression disorder patients. Interestingly, in our study, we found that among the proteins dysregulated in the OXPHOS pathway in WT Stress mice compared to WT Ctrl, 8 out of 15 dysregulated proteins belonged to complex I. In line with this observation, Weger and colleagues found that dysregulation of proteins of complex I indeed better correlated with depression-like phenotype of their stress protocol [43]. Together, this suggests that dysregulation of complex I might be a specific stress-driven pathway ultimately contributing to depression pathology. Proteomics analyses of hippocampal non-synaptic mitochondria extracted from rats following 6 weeks of social isolation revealed dysregulation of metabolic pathways like OXPHOS and TCA cycle, predominantly due to downregulation of involved proteins [44]. This study also found the highest impact of isolation stress on complex I among the OXPHOS-related proteins, which could be reverted by antidepressant fluoxetine treatment together with improvement of the stress-induced effects on OXPHOS and the TCA cycle. Altogether, this indicate that complex I is sensitive to environmental stressors and could be a potential target for drug discovery for stress disorders with symptoms of depression and anxiety.
There was no clear pattern in the dysregulated proteins annotated to OXPHOS pathway in APPPS1 Stress and APPPS1 Ctrl but in the APPPS1 Stress group more proteins of this pathway were dysregulated. This indicates that amyloid pathology drives widespread damage to the respiratory chain and that chronic stress adds to this deficit. Recently, another study from our group found that the change in proteome of senile plaques in Aducanumab-treated APPPS1 mice could be linked to modulations of the OXPHOS, AD and calcium signaling pathways [29]. Specifically, Aducanumab treatment upregulated proteins associated with mitochondrial function. Based on these findings, and the established toxicity of Aβ on mitochondrial function [45], we concluded that the Aducanumab-induced effect on senile plaques was via modulation of cellular pathways associated with Aβ toxicity [29]. Chronic stress has previously been suggested to increase amyloid pathology [4, 46]. Therefore, we speculate that the dysregulations of OXPHOS, AD and Metabolic pathways, observed here, might correspond to stress-acerbated AD-like pathology via mitochondrial damage. This is supported by the significantly upregulated levels of the APP protein in APPPS1 Stress mice compared to APPPS1 Ctrl in our data set (see Supplementary Table 2). Nevertheless, we only saw non-significant increase of plaque load in prefrontal cortex and not in the hippocampus. It has been postulated that the overexpression of the APP in itself cause dysregulation of mitochondrial function independently of Aβ generation [47], which could explain the effect observed here together with the lack of altered amyloid pathology in hippocampus, however, this speculation would need further examination.
Mitochondrial complex I and complex III are the main sources of reactive oxygen species (ROS) production [48] in neurodegenerative diseases like AD [49]. Mitochondrial produced ROS can trigger pro-inflammatory pathways like the NLRP3 inflammasome [50] and thus accelerate AD pathology [51]. Since the electron transport chain of the OXPHOS pathway are compromised with chronic stress in our study, it is likely that ROS driven neuroinflammatory processes were activated in these animals, as has been shown previously [52].
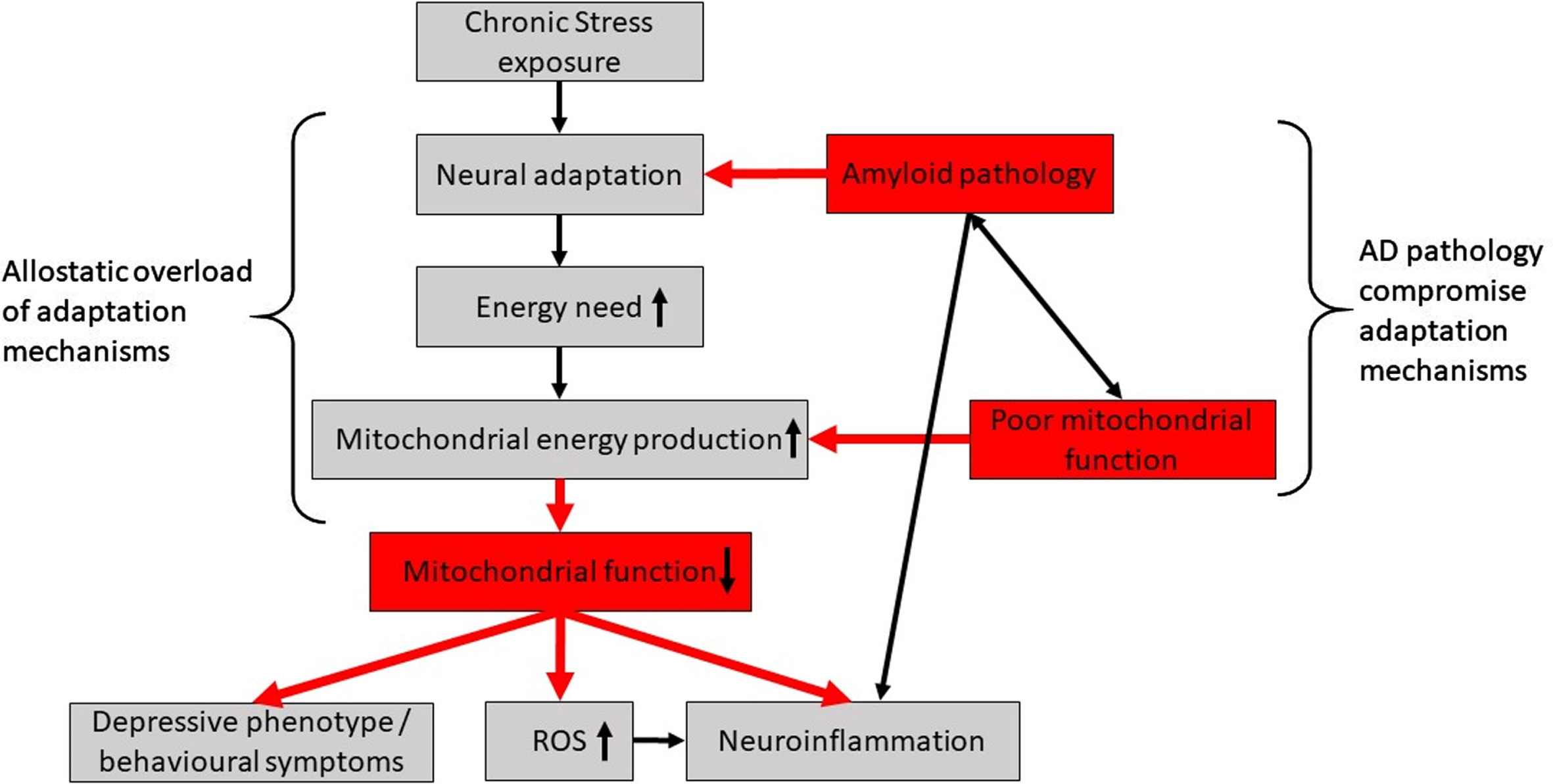
Proposed hypothesis of chronic stress effect on AD brain. To cope with stress exposure, neural networks will adapt and increase energy demand which increase mitochondrial energy production. In AD, these processes are compromised by amyloid pathology burden and poor mitochondrial function. If stress becomes chronic, the adaptive mechanisms will be overloaded leading to reduced mitochondrial function and in turn increase the production of ROS, neuroinflammation and a depressive phenotype or other behavioral symptoms.
Treatment approaches directly targeting mitochondrial damage via antioxidants effects have so far only had limited success in AD [22, 53]. Conversely, some benefit has been shown in preclinical studies [19], though it can be argued that not all preclinical concepts translate to the clinic. One potential reason for the lack of effect in patients might be that the wrong targets have been explored. For example, the antioxidant drugs need to target mitochondrial ROS specifically to induce beneficial effect. Mitoquinone mesylate (MitoQ) is an antioxidant targeting mitochondria that, in preclinical models, prevented AD-like pathology development [54] and progression [55] together with reduced ROS production, astrogliosis, and microglioses. One clinical trial of MitoQ in mild cognitive impairment patients (NCT03514875) is currently recruiting, although the focus is directed towards effects on cerebrovascular blood flow.
Most proteins annotated to OXPHOS and the AD pathway overlapped but 9 proteins of the AD pathway can be linked to synaptic or dendritic functions. CALM1, PPP3R, GNAQ, and MAPK1 were annotated to long term potentiation (LTP), an event involved in synaptic plasticity and memory generation and has been intensively studied in the hippocampus, which is reduced with extreme or chronic stress exposure [7]. CALM1 and GNAQ were downregulated in both APPPS1 Stress and WT Stress compared to WT Ctrl. CALM1 (calmodulin 1) is required for activation of calcium-calmodulin-dependent protein kinase II (CaMKII), which is necessary for LTP (reviewed by [56]). GNAQ (Guanine nucleotide-binding protein G(q) subunit alpha) is involved in neurite outgrowth and its expression has been shown to be decreased in hippocampus of C57BL/6 mice subjected to repeated restraint stress [57]. Together, this suggests that hippocampal synaptic plasticity is compromised with chronic stress in both APPPS1 and WT mice. Moreover, synaptic plasticity is highly energy-demanding and require good mitochondrial function [58]. Thus, we speculate that the potential loss of hippocampal synaptic plasticity in APPPS1 and WT mice subjected to chronic stress may be via the dysregulation of OXPHOS and mitochondrial function.
Proteomic changes observed in the animals subjected to this chronic stress protocol are likely not directly linked to high activation of the HPA axis, as circulating corticosterone levels were comparable in all groups at the time of tissue collection. However, it cannot be ruled out that initial activation of the HPA axis elicits the response we observed. We have previously shown that this chronic stress protocol activates the HPA axis (increases plasma corticosterone levels at day 26) but returns to levels comparable to that of non-stressed littermates after 3 weeks [25]. In this study, we used the same chronic stress protocol and the same strain and age of animals and therefore, we reason that the HPA axis was activated during the chronic stress paradigm and may have induced dysregulations of the hippocampal proteome, which persisted after HPA axis normalization, and may have caused the chronic proteome alterations observed here.
Based on our findings and the discussed literature, we propose a testable hypothesis that chronic stress has a negative additive effect on mitochondrial function, which could initiate the development of NPDs in AD and/or increase the progression rate of both symptoms and disease (Fig. 4).
CONCLUSION
Our data show that chronic stress dysregulates mitochondrial function at protein level in the hippocampus with high impact on complex I in WT mice. Our data also suggests that AD-like pathology has widespread effect on the mitochondrial OXPHOS pathway, and that chronic stress adds to this detrimental effect potentially leading to reduced hippocampal synaptic plasticity. Based on our data and review of the literature, we hypothesize that chronic stress exposure compromises mitochondrial function via higher neuronal energy demand and adaptive mechanisms. In the AD-brain, amyloid pathology and poor mitochondrial function worsens these energy-demanding adaptive mechanisms and loss of mitochondrial function increases ROS and neuroinflammation, which ultimately will worsen behavioral outcome and accelerate development of NPDs. This hypothesis remains to be fully substantiated and the impact of chronic stress on functional regions in the AD brain as well as systemic effects needs to be further investigated.
Footnotes
ACKNOWLEDGMENTS
This work was partly founded by the Innovation Fund Denmark under the grant file number 7038-00103B. We further acknowledge Obelske Family Foundation, Svend Andersen Foundation and Spar Nord Foundation for grants that supported the analytical platform which enabled this study. Likewise, we are grateful to the Danish Agency for Science & Higher Education for the funding to support the Danish National Mass Spectrometry Platform for Functional Proteomics (PRO-MS; Grant no. 5072-00007B). Ditte Bech Laursen (Aalborg University), Anett Ravn Neve and Rie Christensen (H. Lundbeck A/S) are acknowledged for technical assistance.