
Abstract
Background:
Olfactory dysfunction is one of the earliest signs of Alzheimer’s disease (AD), highlighting its potential use as a biomarker for early detection. It has also been linked to progression from mild cognitive impairment (MCI) to dementia.
Objective:
To study olfactory function and its associations with markers of AD brain pathology in non-demented mutation carriers of an autosomal dominant AD (ADAD) mutation and non-carrier family members.
Methods:
We analyzed cross-sectional data from 16 non-demented carriers of the Presenilin1 E280A ADAD mutation (mean age [SD]: 40.1 [5.3], and 19 non-carrier family members (mean age [SD]: 36.0 [5.5]) from Colombia, who completed olfactory and cognitive testing and underwent amyloid and tau positron emission tomography (PET) imaging.
Results:
Worse olfactory identification performance was associated with greater age in mutation carriers (r = –0.52 p = 0.037). In carriers, worse olfactory identification performance was related to worse MMSE scores (r = 0.55, p = 0.024) and CERAD delayed recall (r = 0.63, p = 0.007) and greater cortical amyloid-β (r = –0.53, p = 0.042) and tau pathology burden (entorhinal: r = –0.59, p = 0.016; inferior temporal: r = –0.52, p = 0.038).
Conclusion:
Worse performance on olfactory identification tasks was associated with greater age, a proxy for disease progression in this genetically vulnerable ADAD cohort. In addition, this is the first study to report olfactory dysfunction in ADAD mutation carriers with diagnosis of MCI and its correlation with abnormal accumulation of tau pathology in the entorhinal region. Taken together, our findings suggest that olfactory dysfunction has promise as an early marker of brain pathology and future risk for dementia.
Keywords
INTRODUCTION
Olfactory dysfunction is one of the earliest signs of Alzheimer’s disease (AD), highlighting its potential use as a biomarker for early diagnosis [1, 2]. The most prominent olfactory deficit is odor identification, which involves naming individual odors in a forced choice paradigm. Deficits in odor identification have predicted the transition from mild cognitive impairment (MCI) to AD in clinical and community samples [3]. Furthermore, olfactory dysfunction in cognitively normal individuals has been associated with cognitive decline and neurodegeneration [4]; however, the underlying mechanisms have not been elucidated. Previous studies have investigated the relationship between olfactory dysfunction and specific AD markers, including amyloid-β plaques and tau neurofibrillary tangles. In human autopsy studies, both amyloid-β and tau are found in the olfactory nasal epithelium (containing primary olfactory neurons) [5], the olfactory bulb (containing second-order olfactory neurons), and their projections throughout the olfactory neural network [6, 7]. The sense of smell and episodic memory are closely related based on their common anatomical substrates. For this reason, it is expected that AD pathology in medial temporal lobe regions will impact the ability to identify odors, as well as to store and recollect smell memories [8]. In addition, markers of neurodegeneration such as cortical atrophy in memory-related areas of the brain have been found to correlate with selective deficits in episodic memory of odors [9], and smell identification deficits have been associated with atrophy in the hippocampal and parahippocampus cortex [10, 11]. Testing olfactory function objectively in the clinical setting is easy, quick, accessible, non-invasive, affordable, and reliable. Therefore, early changes in olfactory function could serve as a preclinical marker of AD that can be widely used.
Here we studied members of the Colombian kindred with autosomal dominant AD due to the E280A mutation in the Presenilin-1 (PSEN1) gene. Our group previously showed that carriers of this mutation have elevated cortical amyloid pathology accumulation approximately 15 years before expected symptom onset, followed by entorhinal cortex tau 9 years before symptom onset, and neocortical tau and hippocampal atrophy 6 years before symptoms onset [12]. These mutation carriers are genetically determined to develop early-onset dementia in their forties [13], with a median age of onset for MCI at 44 and dementia at 49. Therefore, this cohort offers a unique opportunity to examine some of the earliest changes in olfactory function and elucidate the relationship between olfaction and memory failure in AD in young individuals with no age-related comorbidities. We aim to examine olfactory function and its association with in vivo brain pathology (amyloid and tau) and cognition in individuals with ADAD. We hypothesized that impairments in olfactory function, specifically smell identification and episodic memory of odors, will be present in pre-symptomatic mutation carriers, years before clinical onset and worse performance in smell identification deficits and abnormalities in episodic odor memory will correlate with levels of AD-related pathology.
MATERIALS AND METHODS
Study design and participants
Baseline olfactory function was assessed in 16 PSEN1 E280A mutation carriers (10 cognitively unimpaired mutation carriers and 6 mutation carriers with MCI, and 19 age and education-matched non-carriers from the same kindred who are enrolled in the Massachusetts General Hospital (MGH) COLBOS (Colombia-Boston) longitudinal biomarker study who traveled to Boston between the years 2018 and 2019. Participants were recruited from the Alzheimer’s Prevention Initiative registry of ADAD, which is a collaborative project among the Neurosciences Group of Antioquia and the Banner Alzheimer’s Institute. The main goal is to provide a source of interested research participants for future clinical studies for AD prevention [14]. Those diagnosed with dementia or with a significant medical, psychiatric, or neurological disorder (e.g., stroke, seizures, substance abuse, and other conditions that affect motor, visuospatial or cognitive abilities) were excluded from this study. Participants and study staff were not informed of the participants’ genetic test results.
The study was approved by both the institutional ethics review boards of the University of Antioquia in Medellin, Colombia and the MGH in Boston, MA. All participants provided written informed consent before participating in any procedures.
Clinical and cognitive assessments
Clinical assessments were performed at the Uni-versity of Antioquia in Medellin, Colombia. Parti-cipants underwent a comprehensive neuropsychological evaluation, including the Mini-Mental State Examination (MMSE) [15], a Spanish version of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) word list test, which has been adapted for this Colombian population [16], and the functional assessment staging test (FAST) [17]. In the CERAD word list delayed recall, participants were asked to recall as many words as they could remember from a previously learned list (10 items) after a 10-min delay. Testing was conducted in Spanish by neuropsychologists or by psychologists trained in neuropsychological assessment. Neurological examinations were performed by a neurologist or by a physician trained in the evaluation of neurodegenerative disorders.
Genotyping
Genomic DNA was extracted from blood by standard protocols and PSEN1 E280A characterization was done at the University of Antioquia using methods previously described [18]. Genomic DNA was amplified with the primers PSEN1-S 5′ AACAGCTCAGGAGAGGAATG 3′ and PSEN1-AS 5′ GATGAGACAAGTNCCNTGAA 3′. We used the restriction enzyme BsmI for restriction fragment length polymorphism analysis. Each participant was classified as a PSEN1 E280A carrier or non-carrier.
PET imaging
All participants in this study traveled from Colombia to Boston (USA) and underwent amyloid and tau PET imaging and MRI at the MGH. As reported previously [12], 11C Pittsburgh compound B (PiB) PET was acquired with an 8.5 to 15 mCi bolus injection followed immediately by a 60-min dynamic acquisition in 69 frames (12×15 s, 57×60 s). [F18] Flortaucipir (FTP) was acquired between 80 and 100 min after a 9.0 to 11.0 mCi bolus injection in 4 separate 5 min frames.
11C PiB PET data were expressed as the distribution volume ratio (DVR) with cerebellar grey as reference tissue; regional time-activity curves were used to compute regional DVRs for each region of interest (ROI) using the Logan graphical method applied to data from 40 to 60 min after injection. 11C PiB retention was assessed using a large cortical ROI aggregate that included bilateral frontal, lateral temporal, and retrosplenial cortices as described previously [12]. [F18] FTP specific binding was expressed in FreeSurfer ROIs as the standardized uptake value ratio (SUVR) to cerebellum, similar to a previous report [19], using cerebellar grey matter as reference. The spatially transformed SUVR PET data was smoothed with an 8mm Gaussian kernel to account for individual anatomic differences. SUVR values were represented graphically on vertices at the pial surface. The two ROIs for FTP tau binding comprised the bilateral entorhinal cortex (EC), as the first location of tau buildup and the inferior temporal cortex, as it represents the best proxy of early tau spreading to neocortex in the PSEN1 E280A kindred [12]. Consistent with previous studies with this same population, PET data were corrected for partial volume effects using the geometric transfer matrix method [20].
For exploratory whole-brain analyses, Spearman correlations were used to assess the relationship between OPID18 scores and vertex-wise amyloid-β and tau PET within carriers. PET images were normalized to standard (MNI) space and projected onto the fsaverage surface, and vertex-wise values were sampled at the midpoint of the gray matter. Partial volume correction was applied using the extended Muller-Gartner method implemented in FreeSurfer [21]. Results were displayed as –log10(p), significant at cluster-wise p < 0.05 (minimum cluster extent = 100 mm∧2), with and without false discovery rate (FDR) correction for multiple comparisons. Correlations and visualizations were performed in R v. 4.0.3; clustering and multiple comparisons corrections were performed using FreeSurfer tools.
Olfactory function testing
Participants underwent the Spanish version of the Aromha olfactory battery [9] that assessed Odor Percept Identification (OPID9 and OPID18), odor discrimination (OD10), and odor memory with the Percepts of Odor Episodic Memory (POEM) tests, as previously described [9]. The odors were from the Living collection (IFF) and self-administered via Whispis (Scentovation). First, participants answered questions that surveyed medical factors that may confound olfactory function (septal deviation, difficulty breathing through one side of nose, history of radiation or chemotherapy, history of nasal surgery, active sinus/upper respiratory infection at the time of testing, known nasal polyps, a history of anaphylaxis to nuts, traumatic or congenital anosmia, current or recent [past 6 months] alcohol or substance dependence, or pregnancy). The Spanish version of the AROMHA olfactory battery evaluates several aspects of olfactory function including odor percept identification, olfactory discrimination, and olfactory memory. Odor percept identification refers to the naming of percepts of recently smelled odors from 4-choices that appear after the participant answers a brief question about the recently presented odor (9 odors in the first OPID9 test and 18 odors in the second OPID18 test, separated by 10 min). Olfactory discrimination requires both working memory of previous odors as well as intact executive verbal functions to classify 2 odors as the same or different (10 trials). Olfactory memory is assessed during OPID18 via the Percepts of Odor Episodic Memory test (POEM) by asking participants “Did you smell the odor in first identification test?” Olfactory memory scores depend on the temporal lobe and hippocampus for retrieval of memory of the odor percept [22]. The OPID9, OPID18, and OD10 tests were scored as proportion correct (0.25 is performing at chance for OPID9, and OPID18; 0.5 is performing at chance of the OD10. The POEM test was scored as the POEM index (items correct –items incorrect)/18), where 0 = chance.
Statistical analyses
We used a non-parametric unpaired Wilcoxon rank-sum test for continuous data and Fisher exact test for categorical data to assess group differences in demographic and clinical variables. In addition, non-parametric Spearman correlations were used to evaluate the association between olfactory variables and clinical and neuroimaging parameters. Specifically, we tested the cross-sectional correlations between olfactory identification with the following variables: mean cortical amyloid-β; regional tau in the inferior temporal and entorhinal cortex; the MMSE total score; and the CERAD word list delayed recall score. We also repeated the cross-sectional correlations with age and education as covariates due to their known associations with memory performance in this cohort. All analyses were performed using R version 3.6.3, with package ppcor [23] for partial correlations.
RESULTS
Demographic and neuropsychological data
We included 16 non-demented carriers of the PSN1 E280A ADAD mutation (mean age [SD]: 40.1 [5.3], 10 females, 6 carriers with MCI) and 19 non-carrier family members (mean age [SD]: 36.0 [5.5], 12 females). Demographic and neuropsychological data are presented in Table 1. Compared to non-carriers, mutation carriers were significantly older (p = 0.041), had fewer years of formal education (p = 0.041), and performed significantly worse than non-carriers in the CERAD word list delayed recall (p = 0.001) and the MMSE (p = 0.004). Cognitively unimpaired mutation carriers (n = 10) and non-carriers did not significantly differ in age (p = 0.55), education (p = 0.39), or sex (p = 1.0). There were not significant differences either in the CERAD word list delayed recall (p = 0.06) or the MMSE (p = 0.14).
Demographic, neuropsychological and neuroimaging data of carriers and non-carriers of PSEN1 E280A
MMSE, Mini-Mental State Examination; FAST, Global Deterioration Scale; SD, standard deviation; OPID, Odor Percept Identification; OD10, Odor Discrimination 10; POEM, Percepts of Odor Episodic Memory; *Chance performance on OPID18 and on POEM index.
Olfactory function performance in carriers and non-carriers
Mutation carriers and non-carriers did not differ in olfactory identification, olfactory discrimination, or episodic memory of odors. We performed subgroup analysis among carriers to compared performance in olfactory function among those in the pre-symp-tomatic stage (n = 10) versus MCI (n = 6). Carriers with MCI performed worse in olfactory identification (OPID 18) than cognitively unimpaired carriers (p = 0.028).
Olfactory function performance and age
Across all carriers, greater age was associated with worse olfactory identification, such that carriers closer to their symptom onset had worse olfactory identification (r = –0.52; p = 0.037) (Fig. 1). There were no significant relationships between age and the olfactory function tests in non-carriers (r = 0.029; p = 0.906).
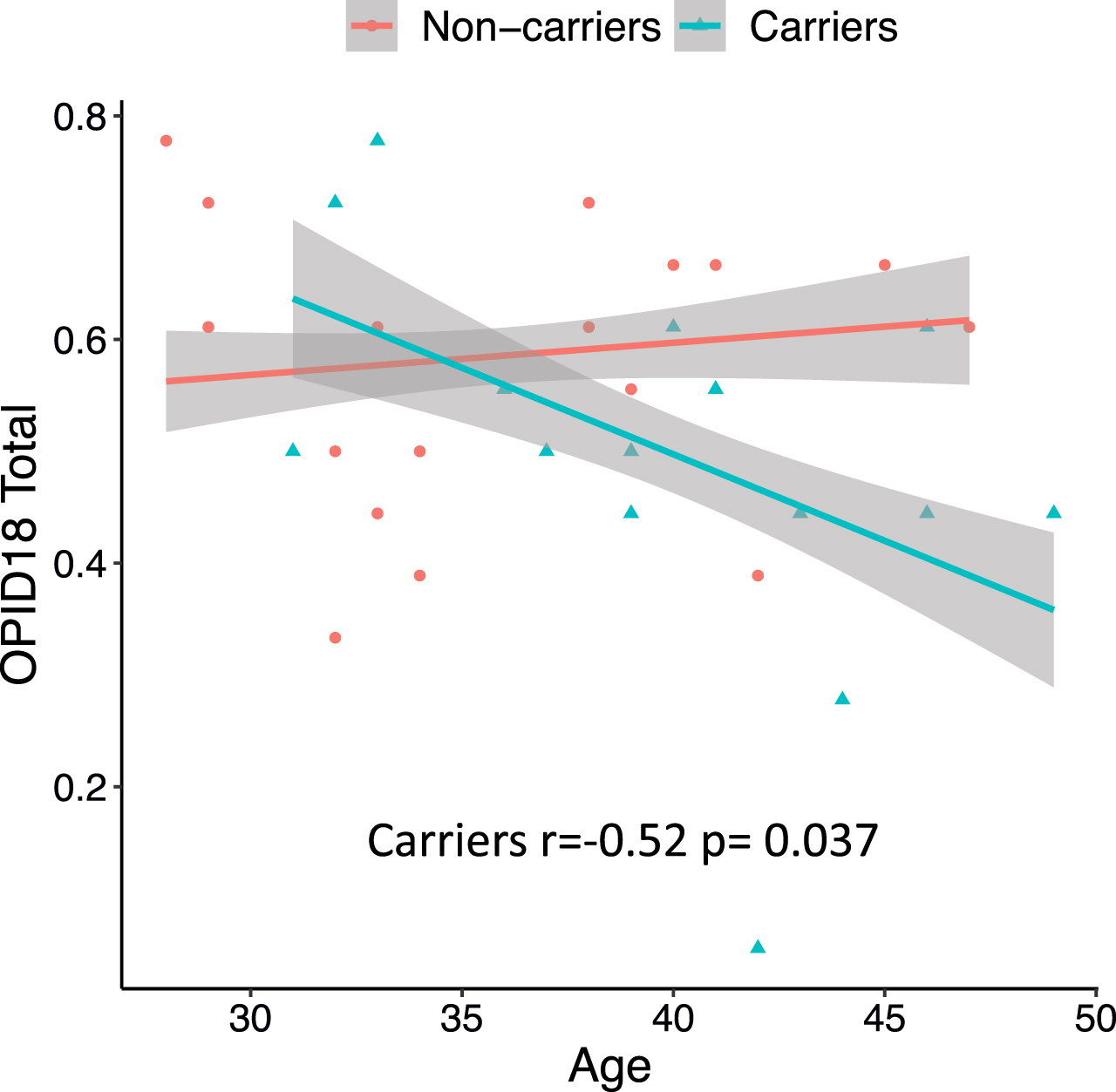
Olfactory function performance and age in mutation carriers and non-carriers. Across all carriers (blue), greater age was associated with worse olfactory identification (OPID18), such that carriers closer to their symptom onset had worse olfactory identification. There were no significant relationships between age and the olfactory function tests in non-carriers (red). Shaded areas represent one standard deviation from the mean.
Olfactory identification (OPID 18) and cognition
In mutation carriers, worse performance in olfactory identification (OPID 18) was related to worse MMSE scores (r = 0.55, p = 0.024) and CERAD delayed recall (r = 0.63, p = 0.007). Adjusting for age and education, there was a trend in the association between OPID 18 and the CERAD word list total correct (r = 0.52, p = 0.055). Odor identification was not associated with cognitive performance in non-carriers. (OPID 18) was not related to worse MMSE scores (r = 0.15, p = 0.543) and CERAD delayed recall (r = 0.06, p = 0.798) in noncarriers.
Olfactory identification (OPID 18) associations with mean cortical amyloid-β and regional tau
Amyloid-β and tau burden for each group are presented in Table 1. Consistent with what our group previously reported [12], all carriers had greater levels of mean cortical amyloid-β (1.66±0.4 DVR; p = 0.001) and regional tau pathology (inferior temporal: 1.44±0.42, p = 0.022; and entorhinal: 1.67±0.79, p = 0.005), compared to non-carriers. In all mutation carriers, worse performance in olfactory identification (OPID 18) was significantly correlated with higher amyloid-β (r = –0.53, p = 0.042), and regional tau pathology (entorhinal: r = –0.59, p = 0.016; inferior temporal: r = –0.52, p = 0.038) (Fig. 2). No associations between OPID 18 and PET markers were observed in non-carriers. In noncarriers, performance in olfactory identification (OPID 18) was not significantly correlated with amyloid-β (r = –0.40, p = 0.088), or regional tau pathology (entorhinal: r = 0.17, p = 0.49; inferior temporal: r = 0.04, p = 0.874).

Olfactory function performance and entorhinal tau in mutation carriers and non-carriers. In all mutation carriers (blue), worse performance in olfactory identification (OPID 18) was significantly correlated with higher regional tau pathology (entorhinal: r = –0.59, p = 0.016). No associations between OPID 18 and tau pathology was observed in non-carriers. Shaded areas represent one standard deviation from the mean.
Olfactory identification (OPID18) and whole-cortex analysis of amyloid-β and tau PET
Spearman correlations were performed between OPID18 and levels of amyloid-β (Fig. 3 left) and tau (Fig. 3 right) burden, as measured by PET within carriers (n = 16). Results are displayed as -log10(p), significant at cluster p < 0.05 before (top) and after false discovery rate (FDR) correction (bottom). Amyloid-β maps suggested involvement of frontal, lateral temporal, retrosplenial, and occipital regions, although no clusters survived FDR correction. Tau maps suggested involvement of temporal and parietal regions, consistent with the known anatomy of early tau accumulation in mutation carriers; only clusters in left medial and lateral parietal cortex survived FDR correction.

Whole-cortex analysis of amyloid-β and tau PET versus olfactory function performance (OPID18). Correlation between OPID18 and amyloid-β (left) and tau (right) PET within carriers. Spearman correlations were performed between OPID18 and amyloid-β (left) and tau (right) PET within carriers (n = 16). Results are displayed as -log10(p), significant at cluster p < 0.05 before (top) and after false discovery rate (FDR) correction (bottom). Amyloid-β maps suggested involvement of frontal, lateral temporal, retrosplenial, and occipital regions, although no clusters survived FDR correction. Tau maps suggested involvement of temporal and parietal regions, consistent with the known anatomy of early tau accumulation in mutation carriers; only clusters in left medial and lateral parietal cortex survived FDR correction.
DISCUSSION
This study examined the associations between olfactory function, in vivo amyloid-β and tau pathology burden, and cognitive performance in non-demented ADAD mutation carriers who will develop dementia with virtually 100% certainty. We found that worse performance in olfactory identification was associated with greater age in carriers, which is a proxy for disease progression in this well-characterized ADAD cohort where mutation carriers have a median age of onset of MCI at age 44 years and dementia at 49 years [13]. This relationship between age and olfactory function was not observed in non-carrier family members. To our knowledge, this is the first study to report olfactory dysfunction in ADAD mutation carriers with diagnosis of MCI and its correlation with abnormal accumulation of tau pathology in the entorhinal region.
Previous studies have shown that olfactory dysfunction increases with age around the 60s in typically aging individuals [8, 24], meaning that age confounds the evaluation of olfactory function in older subjects. Our study, however, evaluated a group of young individuals from an ADAD cohort (mean age non-carriers versus carriers of 36 and 40 years old, respectively) who otherwise did not have risk factors for olfactory dysfunction. We found evidence of worse olfactory function in carriers closer to the age of onset of cognitive symptoms, suggesting a potential impact of AD pathophysiology on olfactory function.
Our findings are consistent with previous work in persons with Down syndrome who also accumulate AD pathology starting at a young age, and revealed increased olfactory dysfunction as a function of age [25]. Our study and previous work suggest that olfactory dysfunction is likely related to underlying AD pathology.
In our study, we did not find differences in olfactory function when we compared all mutation carriers versus non-carriers. However, among carriers, when comparing cognitively unimpaired versus symptomatic carriers with MCI, those at a stage of MCI performed worse in olfactory identification (OPID 18). This is consistent with previous studies in the literature showing deficits in odor identification in MCI stages in patients with AD [26] and olfactory function has been observed to decline over time as AD progresses in severity [22, 27].
A previous study conducted in 18 participants who were members of a family with a known genetic mutation in the presenilin-1 gene [28] revealed that olfactory function was not impaired before the onset of clinical symptoms and was not able to predict which subjects would later develop dementia. However, the authors did not mention if all the participating family members were confirmed mutation carriers.
Both the sense of smell and episodic memory share common neuroanatomical substrates. A previous study showed increased activation of the entorhinal cortex and hippocampus in odor identification tasks [29], similarly these brain regions are activating during verbal/visual episodic memory tasks as well [30]. In mutation carriers, worse performance in olfactory identification (OPID 18) was related to worse performance on clinical and memory measures and showed a trend towards significance after adjusting for age and education. Our findings are consistent with previous studies that have found that olfactory function is related to episodic memory [31, 32]. In addition, earlier reports in the literature have shown that olfactory identification is a strong predictor of memory recall [33] and global cognitive performance [2].
The primary olfactory cortex, particularly the piriform cortex, is close to the entorhinal cortex [34], which is a region that accumulates AD pathology early in the disease process, specifically tau. Therefore, it is expected that AD pathology in medial temporal lobe regions will impact the ability to identify odors, as well as storing and recollecting odor memories. To date, the relationship between amyloid and tau pathology in vivo with performance on olfactory function tests has been limited. We examined regional tau burden in the entorhinal cortex and the inferior temporal cortex since these regions are associated with early tau accumulation in the PSEN1 E280A kindred [12]. We found that in mutation carriers, worse performance in olfactory identification (OPID 18) was related to higher levels of cortical amyloid-β, and tau pathology in the entorhinal and inferior temporal lobe. Our results suggest that AD pathology both (amyloid and tau) are potential drivers of olfactory dysfunction in the absence of other factors that could explain olfactory dysfunction at a young age in this cohort.
In contrast, a previous study found that tau deposition and atrophy in the temporal lobe, but not amyloid deposition, were associated with worse performance on olfactory identification tasks[35] in older individuals with subjective cognitive decline and MCI. In addition, another study found that the absence of olfactory dysfunction predicted a negative amyloid PET scan with a 100% negative predictive value and 41% positive predictive value, suggesting that individuals with normal olfactory function are less likely to have cerebral amyloidosis [36]. Thus, adding olfactory testing to the armamentarium for the clinician could be easy to implement and could be used as a tool to screen for people who may need further workup, along with blood markers when they become available for the clinical use.
The present study has limitations. Our sample size is small compared to other studies of AD and cognitive aging. However, individuals with these mutations are relatively rare. All our participants had a single mutation (PSEN1 E280A), making our sample highly homogeneous compared to other cohorts and one of the larger single mutation ADAD samples with olfactory function tests and PET imaging. More research is also needed to examine whether our findings in ADAD generalize to preclinical late-onset sporadic AD.
Our study has multiple strengths. First, we did not rely on presenting symptoms or cognitive data to infer whether individuals will develop dementia. Instead, we examined olfactory function in a group of individuals with a well-characterized clinical trajectory with MCI starting at a median age of 44 years and dementia at 49 years [13]. Studying ADAD provides a unique opportunity to understand biomarkers of AD in the preclinical stage, as we can estimate how far mutation carriers are from the clinical symptom onset based on the mutation that they carry. Mutation carriers were also young and otherwise healthy, which minimizes potential confounding variables that are more common in advanced age and contribute to olfactory dysfunction. In addition, we examined in vivo amyloid-β and tau pathology using PET imaging, which is considered the gold standard for quantifying and examining in vivo brain pathology in AD.
Taken together, our findings suggest that olfactory dysfunction is associated with greater age in PSEN1 E280A mutation carriers and may be an earlier marker of brain pathology and future risk for dementia. Thus, these results support the potential value of olfactory function test as a helpful tool to assess patients who present with memory complaints. Nevertheless, it should be noted that our findings need to be interpreted with caution given the relatively small sample size. Our findings require further validation with a larger sample size and in individuals with preclinical markers of AD pathology who are carriers of other mutations in autosomal dominant AD and sporadic AD.
Footnotes
ACKNOWLEDGMENTS
The authors thank the Colombian families with autosomal dominant Alzheimer’s disease for contributing their valuable time and effort, without which this study would not have been possible. We also thank Francisco Piedrahita, Alex Navarro, and Claudia Ramos from Grupo de Neurociencias, Universidad de Antioquia in Medellin, Colombia, as well as Jairo Martinez, and Diana Munera from the Massachusetts General Hospital in Boston, MA for helping coordinate visits to Boston and assisting with data collection and processing. We thank International Flavors and Fragrances (IFF) for donating the odors used in this study.
Dr. Ramirez-Gomez was supported by a National Institute on Aging (NIA) Diversity Supplement (P30AG062421-01S1). Dr. M. Albers was supported by the National Institute on Aging NIH R42 AG062130. Mr. Fox-Fuller was supported by the National Institute on Aging (NIA F31AG062158). Dr. Vila-Castelar is supported by a grant from the Alzheimer’s Association (2019A005859). Dr. Lopera was supported by NIH, Roche and an Anonymous Foundation, and the Administrative Department of Science, Technology and Innovation (Colciencias Colombia;111565741185). Dr. Quiroz was supported by grants from the NIH Office of the Director (DP5OD019833), the NIA (R01 AG054671], the Alzheimer’s Association, and Massachusetts General Hospital ECOR.
The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author Y.T.Q. The data are not publicly available because they contain information that could compromise research participant privacy and anonymity.