
Abstract
Background:
NADPH oxidase 2 (NOX2) is an important source of reactive oxygen species (ROS). Activated NOX2 may contribute to Alzheimer’s disease (AD). Our previous studies showed that a novel vitamin E mixture, Tocovid, had potential neuroprotective effects in a stroke mice model and an AD cell model.
Objective:
The aim of this study was two-fold: to assess whether long-term Tocovid treatment can regulate NOX2, and the therapeutic effects of long-term administration of Tocovid to an AD mice model.
Methods:
Therapeutic effects of long-term administration of Tocovid (200 mg/kg /day) on an Aβ-overexpressed transgenic AD mice model (APP23, n = 8) was investigated. The therapeutic effect of Tocovid in 16-month-old mice compared with the no-treatment APP23 group (n = 9) was assessed.
Results:
Tocovid treatment strongly improved motor and memory deficits of APP23 mice by attenuating NOX2 expression, oxidative stress, neuroinflammation, neurovascular unit dysfunction, synaptic alteration, and Aβ deposition after 16 months.
Conclusion:
These findings suggest that NOX2 is a potential target in AD pathology. Long-term administration of Tocovid may be a promising candidate for AD treatment.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by deficits in memory and cognitive function and has also been recognized as the most common cause of dementia in elderly patients [1, 2]. Evidence suggests that elevated oxidative stress in AD brains is caused, at an early stage, by an imbalance between the generation and cleavage of reactive oxygen species (ROS) [3]. NADPH oxidase (NOX) produces ROS and is a fundamental source of ROS in the human body [4]. The NOX family consists of seven ROS-generating enzymes, including NOX1 to 5 and two oxidase isoforms, 1 and 2 [5]. NOX2 is highly expressed throughout components of the central nervous system, including microglia.
Vitamin E is a family of eight naturally occurring homologues, including four tocopherols and four tocotrienols, each of which has an α, β, γ, and δ isoform, whose potent antioxidant capacities are well known. α-Tocopherol has been extensively studied and is considered as one of the most important antioxidants in the brain, whereas research on tocotrienols is limited [6, 7]. A traditional oral vitamin E supplement that only contained α-tocopherol failed to prevent cognitive decline [8]. In contrast, a mixture of tocopherols and tocotrienols had a greater neuroprotective effect [9, 10]. A relatively new vitamin E mixture, Tocovid (α-tocotrienol 12.4%, β-tocotrienol 2.5%, γ-tocotrienol 19.2%, δ-tocotrienol 6.3% and α-tocopherol 11.3%), showed neuroprotective activities in both a stroke mice model and a cell line treated with an amyloid-β (Aβ) oligomer [11, 12].
Despite those findings, the treatment effect of Tocovid against memory and cognitive function deficits in AD remains unknown. Therefore, the present study aimed to assess the therapeutic effects of long-term Tocovid treatment of AD in a mouse AD model. This study also investigated whether Tocovid treatment regulates NOX2 expression in order to evaluate the role of overexpressed NOX2 in AD pathophysiology.
MATERIALS AND METHODS
Animals
All animal experiments were conducted following the guidelines of the Animal Committee of the Graduate School of Medicine and Dentistry of Okayama University (OKU-2022-589). APP23 mice, an AD transgenic mice model overexpressing human Swedish mutant amyloid precursor protein, progressively shows senile plaque and cerebral amyloid angiopathy (CAA). C57BL/6J mice were used as the control. Experimental mice were distributed into three groups: wild type C57BL/6J (WT, n = 10), heterozygous APP23 (APP23, n = 9), and heterozygous APP23 plus Tocovid treatment (APP23+Tocovid, n = 8). From four months (M) of age, Tocovid (200 mg/kg/day) was administered orally though bottle-fed ad libitum after mixing with liquid gel (MediDrop® Sucralose, ClearH2O, Westbrook, ME, USA) [12]. Mice were housed in a 12-h light-dark cycle with controlled temperature around 23°C and ad libitum access to water and food (MF; Oriental Yeast, Tokyo, Japan).
Behavioral analysis
A rotarod performance test was performed to assess balance and motor coordination of mice at the age of 5, 10, and 15 M. To evaluate the riding time that mice could endure on a rotating rod (MK670; Muromachi Kikai Co., Tokyo, Japan), the rotarod performance test started with a rotation speed of 4 rpm, accelerating to a maximum speed of 40 rpm within 60 s. After mice were placed on the rotating rod, the time taken for mice to fall off the rotating rod (up to a maximum of 400 s) was recorded. The best result of three trials was recorded for eachmeasurement.
The eight-arm radial maze test was performed to evaluate behavioral memory, mainly working memory, when mice were 6, 11, and 16 M old [13, 14]. For each trial, mice were released individually into the center of a radial maze and allowed to explore the maze until either all four pellets were consumed or 5 min elapsed. Re-entry of the baited arm which had previously been visited was counted as a working memory error. During training, access to food was restricted for 7 days to gradually reduce weight to 85% of initial weight. Furthermore, mice were allowed to freely explore the maze for 10 min per day before the formal test.
Tissue preparation
After the last behavior test, all mice were sacrificed at 16 M of age. All mice were killed by an overdose of anesthesia and transcranially perfused by ice-cold phosphate-buffered saline (PBS, pH 7.4) followed by 4% ice-cold paraformaldehyde (PFA) in 0.1 mol/L PBS. Brains were removed and post-fixed in 4% PFA overnight. After fixation, brain tissues were separately transferred to ice-cold 10, 20, and 30% (w/v) sucrose in PBS for 24 h. Brains were stored at –80°C after rapid freezing in liquid nitrogen. Frozen brains were sectioned as coronal cryosections (20μm) and placed on silane-coated glass coverslips.
Immunohistochemistry
Double immunofluorescence staining on frozen mice brain sections was performed. To block lipofuscin autofluorescence, sections were pretreated with a TrueBlack lipofuscin autofluorescence quencher (Biotium, San Francisco, CA, USA). Sections were incubated with the following primary antibodies overnight at 4°C: mouse anti-NOX2 antibody (1:100, Santa Cruz Biotechnology, TX, USA), rabbit anti-NeuN antibody (1:200, Abcam, Cambridge, UK), goat anti-CD31 antibody (1:50, R&D Systems, MN, USA), rabbit anti-NG2 antibody (1:500, Sigma-Aldrich, St. Louis, MI, USA), rabbit anti-Iba-1 antibody (1:500, Fujifilm, Osaka, Japan), rabbit anti-PDGF receptor β (PDGFR-β) antibody (1:100, (28E1) Cell Signaling Technology, MA, USA), and mouse anti-GFAP antibody (1:200, Cell Signaling Technology). The antibodies against NOX2, NeuN, CD31, NG2, Iba-1, PDGFR-β, and GFAP were incubated with suitable secondary antibodies conjugated with Alexa Fluor (1:500; Invitrogen, Carlsbad, CA, USA). After mounting with Vectashield mounting medium containing DAPI (Vector Laboratories, Newark, USA), Slides were viewed under a confocal microscope (LSM-780; Zeiss, Jena, Germany). These analyses were conducted by an investigator who was blinded to experimental groupassignment.
For single immunohistochemistry with 3,3′-diaminobenzidine (DAB, Fujifilm), sections were treated with 0.6% hydrogen peroxide/methanol to block the intrinsic activity of peroxidases, followed by 5% bovine serum albumin for 1 h. Sections were incubated with the following primary antibodies overnight at 4°C: mouse anti-NOX2 antibody (1:100, Santa Cruz Biotechnology), mouse anti-4-HNE antibody (1:50; JaICA, Shizuoka, Japan), mouse anti-8-OHdG antibody (1:50; JaICA), goat anti-TNF-α antibody (1:100, R&D Systems), rabbit anti-Iba-1 antibody (1:500, Fujifilm), rabbit anti-fibrinogen antibody (1:100, Abcam), rabbit anti-cleaved caspase 3 antibody (1:100, Cell Signaling Technology), mouse anti-amyloid β 40 (Aβ40) antibody (1:500, Fujifilm), and mouse anti-amyloid β 42 (Aβ42) antibody (1:500, Fujifilm). After washing with PBS, antibodies against NOX2, 4-HNE, 8-OHdG, TNF-α, Iba-1, fibrinogen, cleaved caspase 3, Aβ40, and Aβ42 were separately incubated with secondary antibodies (1:500; Vector Laboratories) followed by an ABC Elite complex (Vector Laboratories) at room temperature for 2 h and 30 min. Slides were viewed under a light microscope (Olympus BX-51, Tokyo, Japan) after visualization with DAB. These analyses were conducted by an investigator who was blinded to experimental group assignment. Pixel intensity of mice sections stained with antibodies was measured from three sections and four randomly selected regions per mice brain. Images were captured at 200×magnification. The area of each selected region was 0.145 mm2.
Statistical analysis
Statistical analysis was performed in GraphPad Prism (version 8.3, GraphPad Software Inc., San Diego, CA, USA). All values are expressed as the mean±median (interquartile range). Prior to data analysis, data normality was determined by a Shapiro– Wilk’s test. All data was normally distributed. Comparison of treatment means involved a one-way analysis of variance (ANOVA) followed by the Tukey-Kramer test. p < 0.05 was considered as statistically significant.
RESULTS
Tocovid improved motor and memory deficits
The result of the rotarod test showed that 5, 10, and 15 M old mice of the APP23 group showed significantly greater motor deficits than mice in the WT group (Fig. 1A, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 versus WT, Tukey’s multiple comparison test). Motor dysfunctions were rescued by Tocovid treatment compared with the APP23 group at 15 M (Fig. 1A, #p < 0.05 versus APP23).
The eight-arm radial maze test was used to evaluate the impairment of working memory. At 6, 11, and 16 M of age, a significant increase in error times was observed in the APP23 group compared to the WT group (Fig. 1B, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 versus WT). At 16 M, Tocovid treatment rescued this working memory deficit in errors compared with the APP23 group (Fig. 1B, #p < 0.05 and ##p < 0.01 versus APP23).
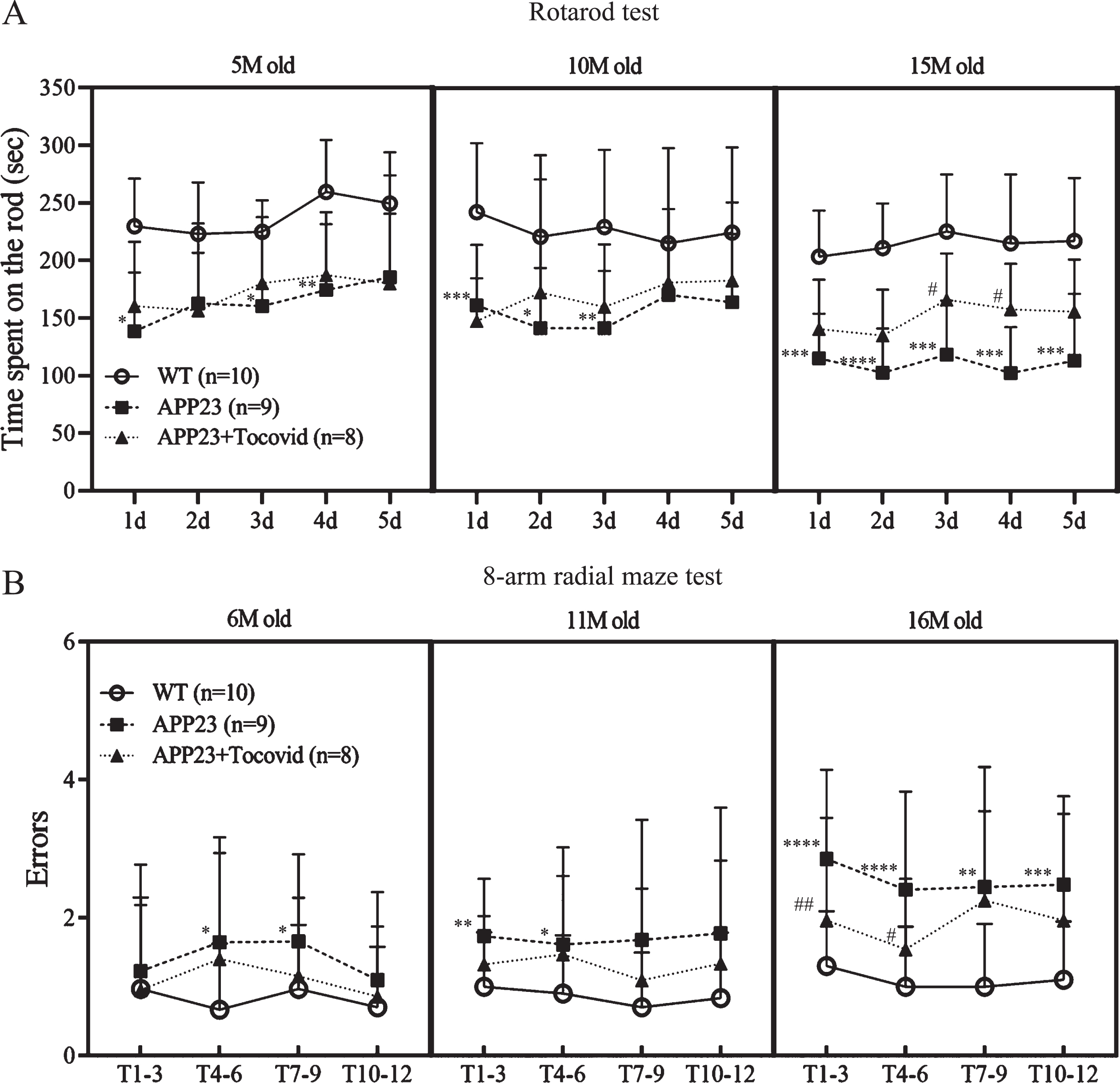
Quantitative analysis of motor function in the rotarod test (A) and working memory by an eight-arm radial maze test (B). Tocovid significantly improved both motor function and memory deficits of the AD mice model (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 versus WT; #p < 0.05 and ##p < 0.01 versus APP23). T1-3, trial1-3; T4-6, trial4-6; T10-12, trial10-12.
Tocovid suppressed NOX2 expression
Double immunofluorescence staining showed that NOX2 was weakly colocalized with some NeuN-positive neurons in the cytoplasm (Fig. 2A). NOX2 was also colocalized with some Iba-1– positive microglia around Aβ plaques, blood vessels, and some NG2-positive oligodendrocyte precursor cells (Fig. 2B–D). However, NOX2 was not colocalized with CD31-positive endothelial cells, PDGFR-β-positive pericyte cells (Fig. 2E, F), or GFAP-positive astrocytes (Supplementary Figure 1A). The results of single immunohistochemistry showed significantly higher NOX2 expression in the APP23 group than in the WT group in 16 M old mice in both the cortex and hippocampus (Fig. 2G, H; **p < 0.01 and ****p < 0.0001 versus WT). However, Tocovid treatment strongly inhibited the accumulation of NOX2 in both the cortex and hippocampus compared to the APP23 group (Fig. 2G, H; ##p < 0.01 and ###p < 0.001 versus APP23).
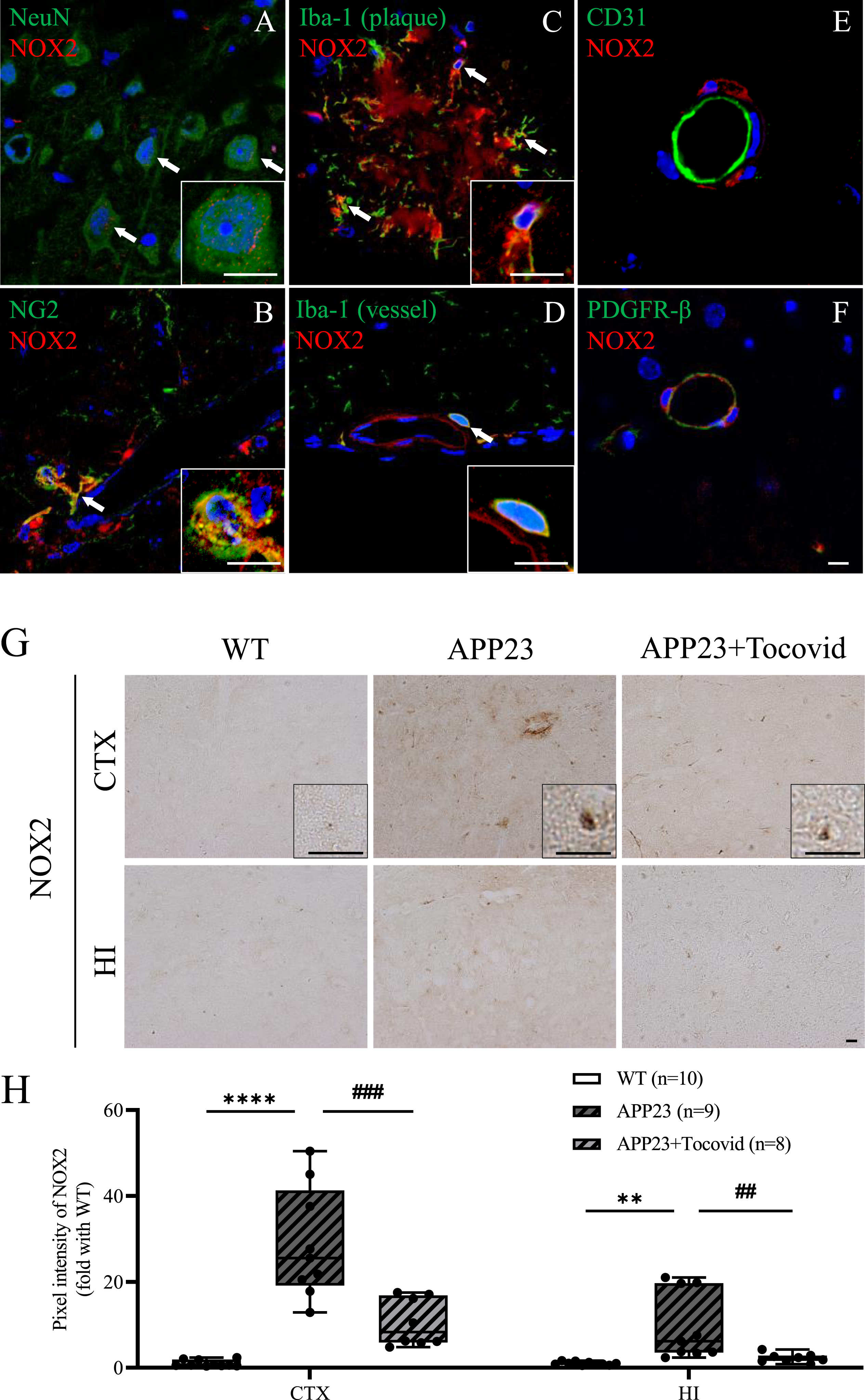
A-F) Double immunostaining of NOX2 with NeuN, NG2, Iba-1, CD31, and PDGFR-β in the cortex of mice in the APP23 group. Scale bar = 10μm. The NOX2 colocalized with NeuN, NG2, and Iba-1-positive cells (arrows), but not with CD31 or PDGFR-β. G) Immunochemical staining of NOX2 in the cortex and hippocampus of mice model. Scale bar = 20μm. H) Quantitative analysis of NOX2. Note the significant decrease of NOX2 expression in the APP23+Tocovid group compared to the APP23 group (**p < 0.01 and ****p < 0.0001 versus WT; ##p < 0.01 and ###p < 0.001 versus APP23). CTX, cortex; HI, hippocampus.
Tocovid attenuated oxidative stress and neuroinflammation
Compared to the WT group, a significantly higher pixel intensity of a lipid peroxidation marker (4-HNE) and a DNA oxidation marker (8-OHdG) was observed in both the cortex and hippocampus of 16 M old mice of the APP23 group (Fig. 3A–D; ****p < 0.0001 versus WT). However, Tocovid treatment greatly attenuated these oxidative stress related-markers in the cortex and hippocampus compared with the APP23 group (Fig. 3A–D; #p < 0.05, ##p<0.01, and ####p < 0.0001 versusAPP23).

A, B) Immunochemical staining of 4-HNE and 8-OHdG in the cortex and hippocampus of the mouse model. Scale bar = 20μm. C, D) Quantitative analysis of 4-HNE and 8-OHdG. Note the significant decrease of 4-HNE and 8-OHdG expression in the APP23 + Tocovid group compared to the APP23 group (****p < 0.0001 versus WT; #p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001 versus APP23).
The expression of a microglia marker (Iba-1) increased significantly in the cortex while the expression of an inflammation cytokine marker (TNF-α) increased significantly in both the cortex and hippocampus of the APP23 group, compared to the WT group (Fig. 4A–D; ****p < 0.0001 versus WT). The increase in activated microglia was observed mainly around Aβ plaques in the APP23 and APP23+Tocovid groups (Fig. 4A). However, Tocovid treatment greatly reduced microglia activation in the cortex compared with the APP23 group (Fig. 4A, C; #p < 0.05 versus APP23). Moreover, Tocovid treatment significantly attenuated the expression of TNF-α in both the hippocampus and cortex (Fig. 4B, D; ##p < 0.01 and ###p < 0.001 versus APP23).

A, B) Immunochemical staining of Iba-1 and TNF-α in the cortex and hippocampus of the mouse model. Scale bar = 20μm. C, D) Quantitative analysis of Iba-1 and TNF-α. Note the significant decrease of Iba-1 expression around Aβ plaque (black mark) in the cortex of the APP23+Tocovid group compared to the APP23 group as well as the significant decrease of TNF-α expression in the cortex and hippocampus of the APP23+Tocovid group compared to the APP23 group (****p < 0.0001 versus WT; #p < 0.05, #p < 0.05, ##p < 0.01, and ###p < 0.001 versus APP23).
Tocovid suppressed activation of Caspase 3 around Aβ plaque
The expression of cleaved caspase 3 was mainly around Aβ plaques in the brains of APP23 mice (Fig. 5A). When mice were 16 M old, cleaved caspase 3 expression increased significantly in the APP23 group compared with the WT group in the cortex (Fig. 5A, C; ****p < 0.0001 versus WT). However, Tocovid treatment strongly suppressed the accumulation of cleaved caspase 3 compared with the APP23 group (Fig. 5A, C; ##p < 0.01 versus APP23).
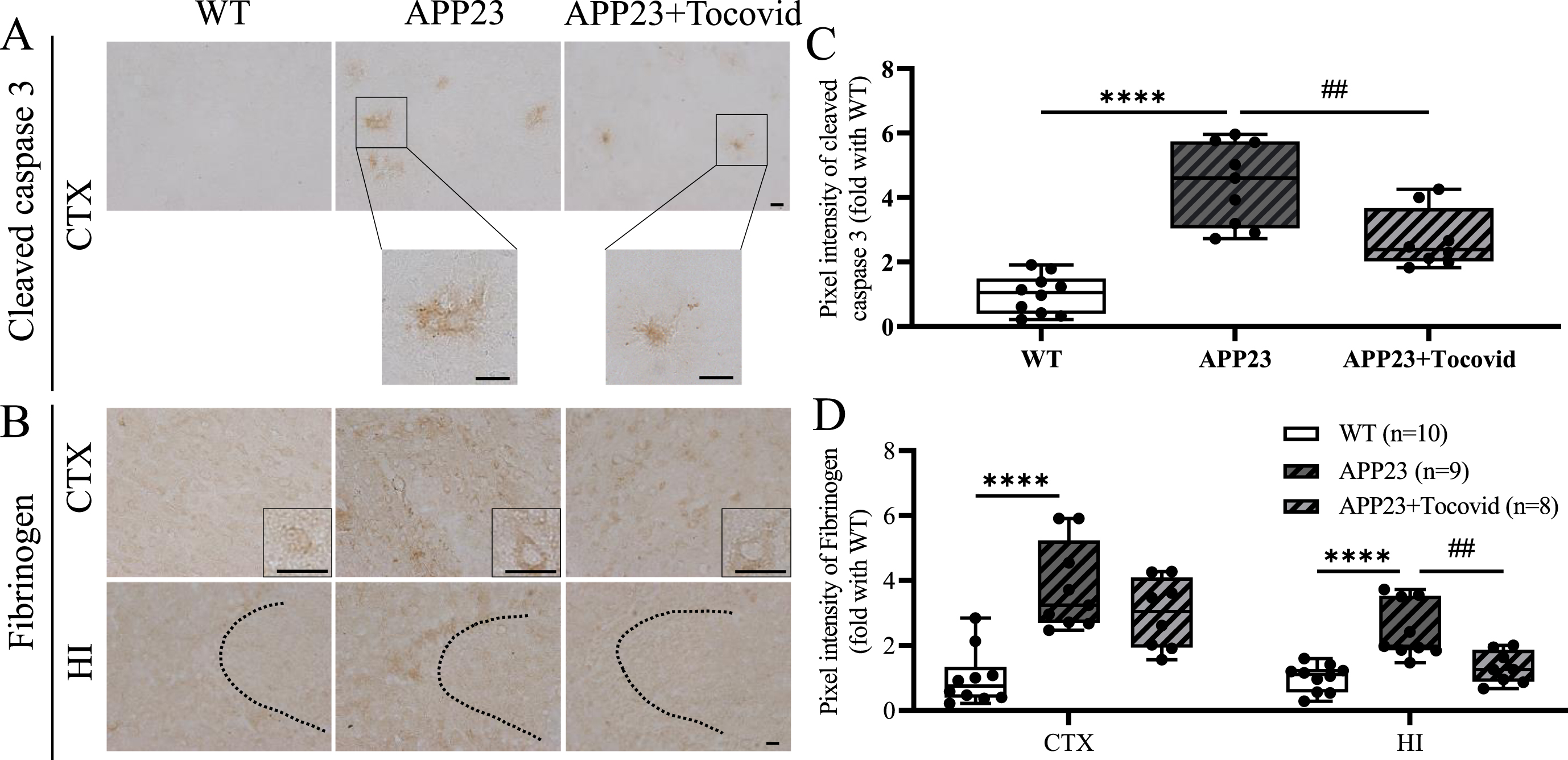
A, B) Immunochemical staining of cleaved caspase 3 in the cortex, fibrinogen in the cortex and hippocampus. Scale bar = 20μm. C, D) Quantitative analysis of cleaved caspase 3 and fibrinogen (****p < 0.0001 versus WT; ##p < 0.01 versus APP23).
Tocovid improved both blood-brain barrier leakage and endothelium/astrocyte remodeling
To evaluating the level of blood-brain barrier (BBB) leakage, the deposition of fibrinogen in the cortex and hippocampus was examined. Both the cortex and hippocampus of the APP23 group showed a significant increase in the expression of fibrinogen in 16 M old mice (Fig. 5B, D; ****p < 0.0001 versus WT). Tocovid treatment attenuated this fibrinogen deposition compared with the APP23 group in the hippocampus (Fig. 5B, D; ##p < 0.01 versusAPP23).
Double immunofluorescent of CD31 and astrocyte foot processes (GFAP) showed that the inner diameter and the area of CD31-positive vessels of a part of small vessels increased in the APP23 group compared to the WT group in 16 M old mice (Fig. 6A–C; ****p < 0.0001 versus WT). Tocovid treatment significantly reduced the inner diameter and CD31-positive area, compared to the APP23 group (Fig. 6A–C; #p < 0.05 and ####p < 0.0001 versusAPP23).
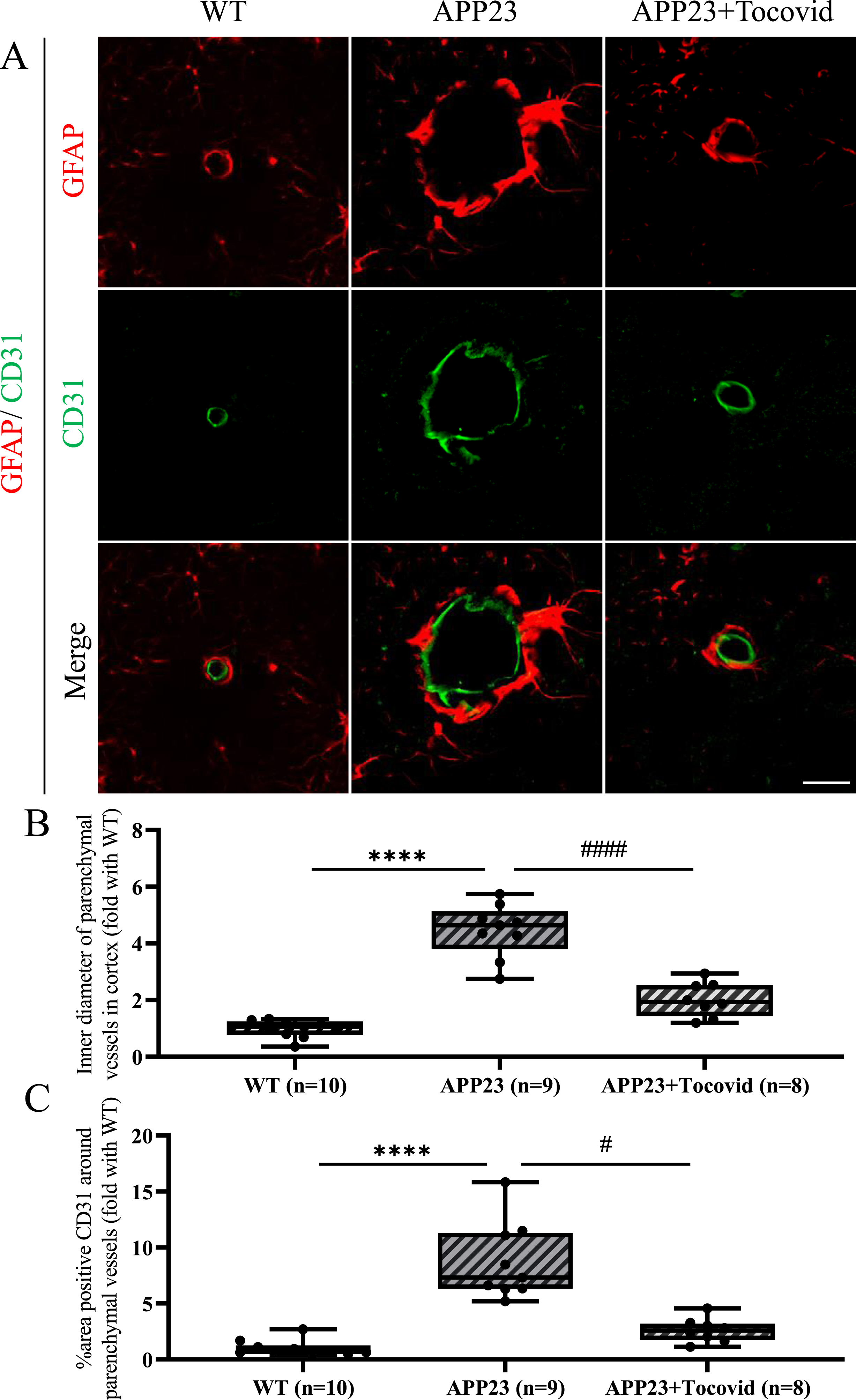
A) Double immunostaining of GFAP and CD31 in the mouse model. Scale bar = 20μm. B, C) Quantitative analysis of inner diameter of parenchymal vessels and area of CD31-positive vessels. Note the significant decrease of inner diameter of parenchymal vessels and area of CD31-positive vessels in the APP23+Tocovid group compared to the APP23 group. (****p < 0.0001 versus WT; #p < 0.05 and ####p < 0.0001 versus APP23).
Tocovid decreased parenchymal and vascular Aβ deposition
When mice were 16 M old, there was an abnormal accumulation of Aβ42 and Aβ40, which increased significantly in the APP23 group compared with the WT group in both the cortex and hippocampus (Fig. 7A–D; ****p < 0.0001 versus WT). The Tocovid-treated group showed a significant decrease in the amount of abnormal deposition of Aβ42 and Aβ40 in the cortex compared with the APP23 group (Fig. 7A–D; #p < 0.05 versus APP23+Tocovid). Moreover, Aβ40 deposition in the hippocampus was attenuated by Tocovid treatment (Fig. 7B, D; #p < 0.05 versus APP23).
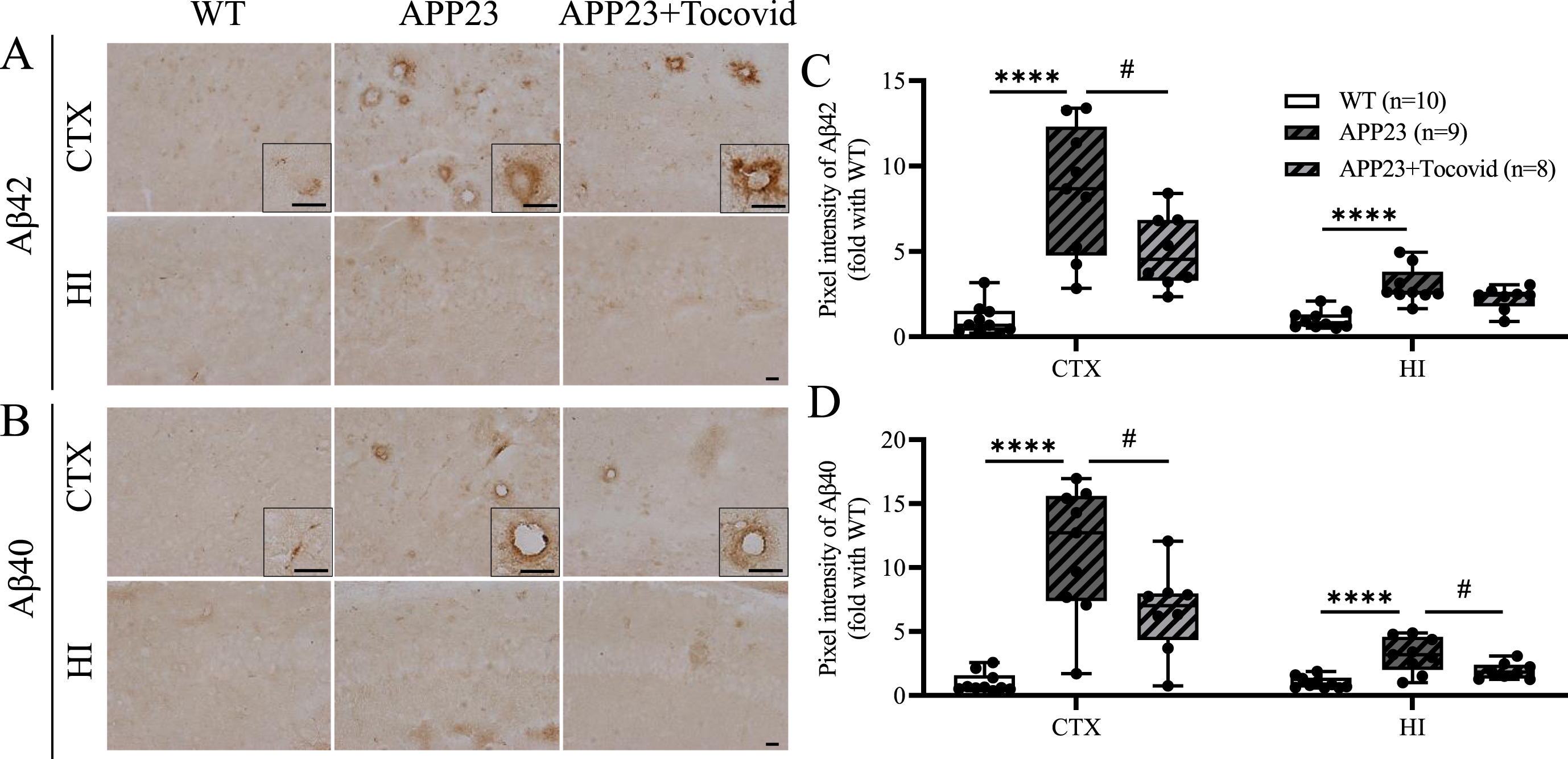
A, B) Immunochemical staining of Aβ42 and Aβ40 in the cortex and hippocampus of the mouse model. Scale bar = 20μm. C, D) Quantitative analysis of Aβ42 and Aβ40 (****p < 0.0001 versus WT; #p < 0.05 versus APP23).
DISSCUSION
In the present study, a long-term treatment of Tocovid, which is a mixture of α-tocopherol and tocotrienols, was applied to APP23 mice between 4 M and 16 M of age to evaluate the therapeutic effects of Tocovid in an AD mice model. Tocovid treatment attenuated motor deficits as well as memory decline in 16 M old APP23 mice (Fig. 1). Moreover, Tocovid treatment partially rescued BBB leakage, oxidative stress, neuroinflammation, and Aβ deposition in the brains of mice (Figs. 3–7).
In the present study, Tocovid showed the ability to inhibit NOX2 expression (Fig. 2G, H). NOX2 is a known important source of ROS, and the excessive activation of NOX2 can easily cause an imbalance in ROS and lead to oxidative stress [15, 16]. Previous studies firmly established that NOX2 expression increased in an aging AD animal model [16–19]. Moreover, recent studies suggested that NOX2 activation could be induced by Aβ42 in both microglia and neurons, thereby causing oxidative stress and inhibiting brain glucose utilization, further leading to Aβ generation [20–22]. In addition, abnormal Aβ accumulation can activate NOX in neuronal and microglial cells, and in the vasculature [21, 23]. These findings indicate that NOX2 overexpression is a potential cause of AD.
Oxidative stress is also a hallmark of AD, which is caused by an imbalance between the generation and cleavage of ROS, and this occurs as an early event in AD [3]. Lipid peroxidation and DNA oxidation production as a result of oxidative stress caused by excessive ROS production, decreased after Tocovid treatment, indicating that oxidative stress was attenuated by Tocovid (Fig. 3) [24, 25]. These anti-oxidative stress effects might be caused not only by neutralizing ROS, but also by inhibiting NOX2 expression [26, 27]. Tocovid also showed an anti-inflammatory effect by suppressing microglia activation, consequently leading to a decrease in the generation of TNF-α, and showing a similar tendency as microglia activation (Fig. 4) [28].
In the present study, cleaved caspase 3 was mainly observed around the senile plaque in APP23 mice but was absent in WT mice (Fig. 5A). This indicates that the increase in cleaved caspase 3 may be caused by abnormal Aβ plaques. Previous studies suggested that activation of Aβ-induced NMDA receptor (NMDAR) could stimulate cleaved caspase 3 and contribute to synaptic alterations [29–33]. Tocotrienols suppressed glutamate receptor1 (GluN1) expression in neural cells in vitro, probably leading to less cleaved caspase 3 expression [34]. These lines of evidence suggest that Tocovid may attenuate synaptic alterations by reducing cleaved caspase 3 expression (Fig. 5A, C).
In current clinical studies, α-tocopherol was unable to show a significant therapeutic effect in AD patients [8]. However, there are limited studies of the therapeutic and preventive effects of tocotrienols in AD patients. In the present study, we demonstrated that Tocovid could rescue memory deficits in an AD animal model, which indicated that Tocovid might have beneficial effects in AD patients. Our findings provide supporting evidence for applying clinical experiments of Tocovid treatment in AD patients in the future.
In summary, the present study demonstrated that Tocovid was able to attenuate motor and memory deficits, oxidative stress, neuroinflammation, neurovascular unit dysfunction, synaptic alteration and Aβ deposition in the APP23 mice model. These findings provide evidence that NOX2 may be a potential target in AD pathology, breaking the vicious cycle of NOX2 and Aβ deposition in APP23 mice, a state that can be partially reversed by Tocovid. Therefore, Tocovid showed obvious therapeutic effects in this AD mice model and may be a promising candidate for AD treatment.
Footnotes
ACKNOWLEDGMENTS
The authors are grateful to Alan Foo (Hovid, Malaysia) for the kind gift of Tocovid.
FUNDING
This study was partly supported by a Grant-in-Aid for Scientific Research (C) 20K09370, 20K12044, Challenging Research 21K19572, Young Research 20K19666, 21K15190, and by Grants-in-Aid from the Research Committees (Toba K, and Tsuji S) from the Japan Agency for Medical Research and Development.
CONFLICT OF INTEREST
Koji Abe and Toru Yamashita are Editorial Board members of this journal but were not involved in the peer-review process nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and its supplementary material.