
Abstract
Alzheimer’s disease (AD) is a pathological disorder defined by the symptoms of memory loss and deterioration of cognitive abilities over time. Although the etiology is complex, it is mainly associated with the accumulation of toxic amyloid-β peptide (Aβ) aggregates and tau protein-induced neurofibrillary tangles (NFTs). Even now, creating non-invasive, sensitive, specific, and cost-effective diagnostic methods for AD remains challenging. Over the past few decades, polymers, and nanomaterials (e.g., nanodiamonds, nanogold, quantum dots) have become attractive and practical tools in nanomedicine for diagnosis and treatment. This review focuses on current developments in sensing methods such as enzyme-linked immunosorbent assay (ELISA) and surface-enhanced Raman scattering (SERS) to boost the sensitivity in detecting related biomarkers for AD. In addition, optical analysis platforms such as ELISA and SERS have found increasing popularity among researchers due to their excellent sensitivity and specificity, which may go as low as the femtomolar range. While ELISA offers easy technological usage and high throughput, SERS has the advantages of improved mobility, simple electrical equipment integration, and lower cost. Both portable optical sensing techniques are highly superior in terms of sensitivity, specificity, human application, and practicality, enabling the early identification of AD biomarkers.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a deadly neurological disorder that mostly affects the elders. It has a high morbidity rate caused by the damages on the brain to the point of causing memory loss and exacerbates cognitive problems over time [1]. After cancer, heart disease, and cerebrovascular illness, AD ranks as the fourth most common cause of death for older individuals [2, 3]. A greater than 145% rise in AD fatalities occurred between 2000 and 2019 [4]. According to Dementia Forecasting Collaborators, 153 million people worldwide will be affected by AD by 2050 [5]. It is estimated that there will be over one million new cases annually, or one new case every 33 seconds [6]. Patients can experience preclinical AD symptoms like mild cognitive and memory impairment, and eventually dementia and frailty as AD progresses [7, 8].
For the standard AD diagnosis, practitioners have a few well-known biomarkers in their arsenal [9–11]. Tau neurofibrillary tangles and amyloid-β (Aβ) plaques are AD’s two most common pathological indicators [12, 13]. Positron emission tomography (PET) imaging and cerebrospinal fluid (CSF) examinations employing immunoassays that assess Aβ42 (or Aβ42/Aβ40 ratio), total-tau (t-tau), and phosphorylated tau (p-tau) are both capable of detecting these disorders in living persons [14]. The other predictive biomarkers are neurofilament light (NfL), flotillin, alpha-1 antitrypsin (AAT), thrombin, amyloid-β protein precursor (AβPP), Alzheimer-associated neuronal thread protein (AD7c-NTP), thiamine pyrophosphate, β-site APP-cleaving enzyme (BACE1), and Aβ -derived diffusible ligands (ADDLs) [15, 16].
A decade before symptoms emerge, neuropathological alterations may have occurred [17]. Late intervention is a significant contributor to treatment failure, according to growing evidence from clinical studies on the risk factors for AD [18]. In the patient’s brain (Fig. 1a, the blue shaded area of the left panel), plaques and nerve fiber tangles gradually spread to other cortical regions in a predictable pattern. In early AD, the brain undergoes the development of plaques and tangles in regions responsible for cognitive processes such as learning, thinking, and memory, even before the symptoms are identified with existing testing methods. In the moderate stage, plaques and tangles expand to where the patient finds speaking, understanding speech, and recognizing objects to be complex tasks. Most of the cerebral cortex suffers severe damage in the advanced stages of AD. The brain also undergoes significant atrophy due to the widespread loss of brain cells. Patients lose their capacity for self-care, self-identification, and communication. The typical patient lives for around eight years. However, the propagation speed of the disease is correlated with the age of diagnosis and the patient’s physical condition at the time, therefore some patients can live for up to 20 years [19]. Thus, detecting AD in its early stages, prior to the onset of symptoms, can be very beneficial in the prevention and management of the disease, which currently has no cure [20].

According to Fig. 1b [11], the earliest noticeable biomarker shifts mimicking AD pathology is a decrease in CSF Aβ levels, followed shortly by PET amyloid imaging. Even before dementia develops, these two amyloid pathology markers may reach their peak pathogenic levels. When looking at individuals with cognitive impairment, it is important to consider how biomarkers for AD evolve over time. Although biomarkers such as CSF tau, fludeoxyglucose-PET, and structural brain imaging indicate neuronal degeneration at a slower rate than amyloid biomarkers, the progression of cognitive decline is not a linear process due to the influence of both genetic and environmental factors. Thus, when interpreting test results for people with cognitive impairment, it’s important to take into account the temporal changes in AD biomarkers [21].
A primary goal is to create cost-effective, non-invasive, and sensitive diagnostic techniques for AD. Intending to diagnose AD accurately, researchers have undertaken impressive efforts to develop trustworthy and time-efficient technologies for the quantitative and selective measurement of the disease’s biomarkers. Their efforts have led to clinically useful photoelectric assessment techniques with great sensitivity, excellent cost performance, repeatability, and ease of use. Novel optoelectronic sensing systems provide a quick, simple, and affordable way of analysis compared to traditional techniques such as gas chromatography (GC), high-performance liquid chromatography (HPLC), and mass spectrometry (MS) [22]. Our review will now summarize the development of useful optical analytic technologies for the identification of AD biomarkers, as shown in Fig. 2.
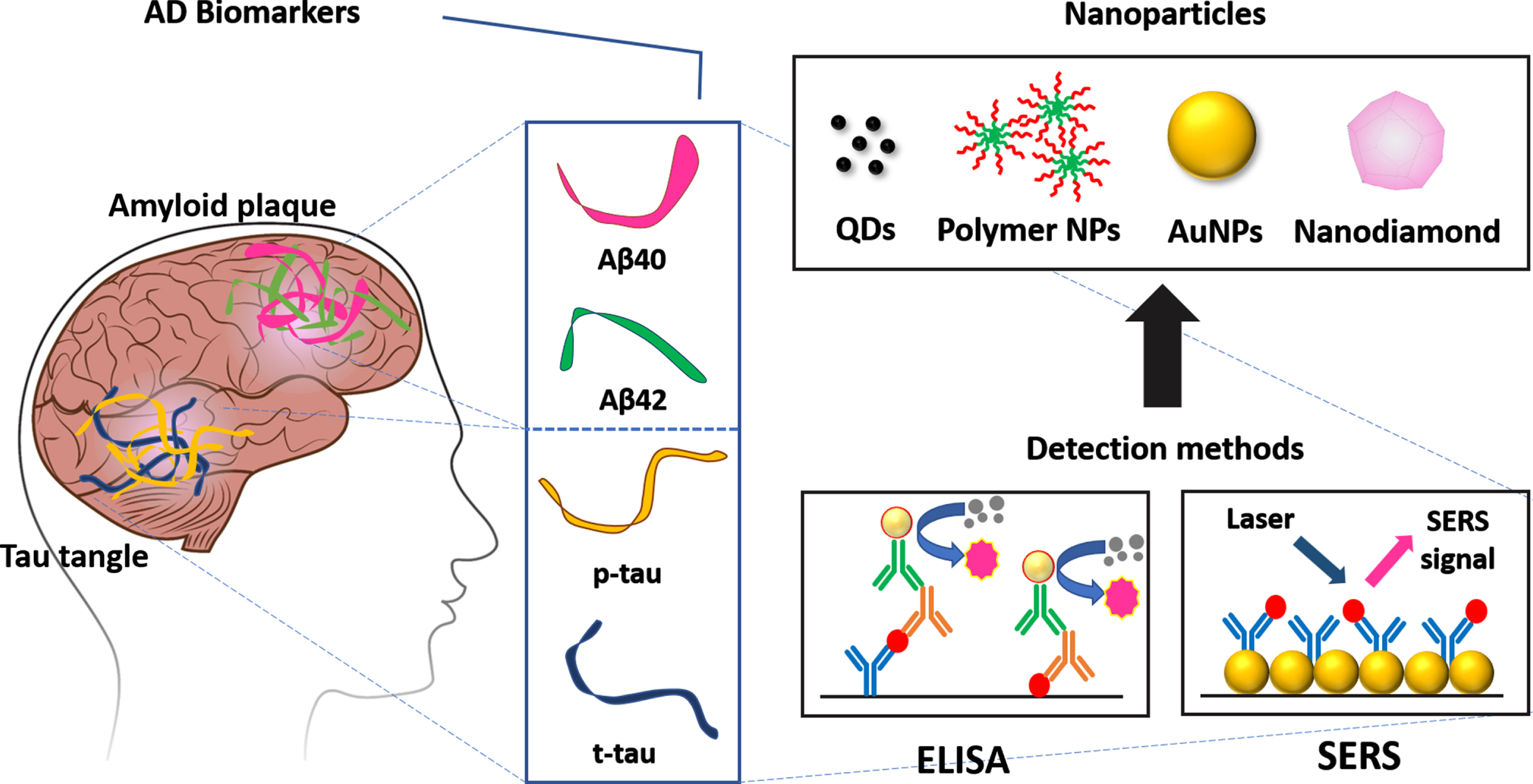
The development of detection methods for Alzheimer’s disease (AD).
In particular, protein analysis by Surface-enhanced Raman spectroscopy (SERS) is efficient. SERS can simultaneously evaluate modifications in structure and concentrations of specific target molecules. Several research areas, including early diagnosis, medication administration, and clinical therapy, are handled by SERS [23, 24]. SERS holds the potential to achieve greater sensitivity compared to alternative techniques, like enzyme-linked immunosorbent assay (ELISA), particularly when the test sample contains minimal quantities of the components being tested [25–28]. Extensive utilization of SERS has been carried out to detect chemical and biological substances qualitatively and quantitatively [25, 29–31]. Despite its potential, practical implementation of SERS is impeded by various challenges in research, such as the irregular coating of analytes, nonuniformity of plasmonic nanostructures, and denaturation of proteins [23]. Quick marker identification is necessary for targeted and efficient disease therapy.
ALZHEIMER’S DISEASE BIOMARKER
In vitro and in vivo physiological, biochemical, and anatomical measurements are known as biomarkers. Biomarkers can be used to identify particular characteristics of pathological changes brought on by illnesses [32]. Several biomarkers, including the tau protein that causes neurodegeneration and the Aβ peptide in cerebral amyloid deposition, have been suggested to have particular roles in the onset of AD. In this section, we will summarize the characteristics of core AD biomarkers in CSF and human blood—namely, the Aβ peptide and tau protein involved in forming senile plaques and NFTs, respectively. We will also discuss other AD biomarkers, such as ApoE and miRNA.
Cerebrospinal fluid proteins
The brain and the spinal cord are covered and cushioned by CSF, a transparent mixture of water, low-concentration proteins, ions, and glucose. Adults typically have one pint of CSF. Doctors can take a sample of CSF using a minimally invasive technique called a lumbar puncture. The levels of many indicators within CSF, including those of tau and Aβ, two factors that cause abnormal brain deposits strongly linked with AD, may fluctuate in the early stages of AD. Another potential marker is NfL, whose quantity has been shown to increase in neurodegenerative diseases such as AD [33].
Aβ40 and Aβ42 peptides
The major clinical characteristic of AD and the primary pathogenic event in the disorder have been postulated to be extracellular deposition of Aβ, that is created through the degradation of AβPP by BACE1 and γ-secretase, resulting in the formation of plaques [34]. Aβ42, 42 amino acids long and susceptible to the aggregation form of Aβ, has decreased concentrations in AD CSF, about 50% lower than those of normal levels [35]. Normally, Aβ42, which is produced when AβPP is broken down, is transported through the glymphatic system from the interstitial fluid of the brain to the CSF and bloodstream [36]. Reduced CSF levels are the result of Aβ42 aggregation in the brain parenchyma.
A valuable diagnostic biomarker of AD, the Aβ42/40 ratio, represents the Aβ42 concentration in CSF relative to Aβ40 concentration [37]. When aggregation-prone Aβ42 is divided by soluble Aβ40, diagnostic accuracy for Aβ pathology improves. Aβ42 and Aβ40 both originate from the same processing line of AβPP. However, Aβ40 stays soluble in AD. Nearly all discordant incidents that are initially positive for CSF but negative for PET tend to become PET-positive within a few years [37–39]. The CSF ratio of Aβ42 to Aβ40, which distinguishes between individuals with high and low Aβ production due to differences in AβPP processing, is strongly associated with amyloid PET [38].
The presence of insoluble clumps of amyloid protein is a pathological hallmark of AD. Aβ protein that has been aggregated makes amyloid plaques, which the brain cortex has all over. Additionally, Aβ aggregates can lead to synaptic loss and neurotoxicity, that will interfere with complicated learning processes, memory, and impede long-term neural potentiation [39].
Tau
Tau, p-tau, and t-tau protein are biomarkers of neuronal injury. The combined Aβ42, p-tau, and t-tau for diagnosing AD yields 85–95% sensitivity and specificity.
The MAPT gene located on chromosome 17 is responsible for the production of the hydrophilic tau protein, which contains sizeable natively unfolded segments and is abundant in the axons of neurons during their development and maturity [40]. The adult human brain contains six variants of tau protein generated by the MAPT gene, which differ in length and consist of 352 to 441 amino acids, with the longer isoform predominating in peripheral organs [41, 42]. The tau protein is composed of four parts: an N-terminal region, a microtubule-binding region, a mid-region, and a carboxy terminus. The N-terminal region of the tau protein, ranging from amino acids 45 to 103, may have zero, one, or two inserts due to alternative splicing, leading to the production of 0N, 1N, and 2N tau, respectively. The microtubule-binding section of the protein (R1–R4), depicted in Fig. 3, consists of three or four pseudo-repeat domains. In the mid-region, there are several threonine and serine residues, and their phosphorylation by certain kinases can have both physiological and pathological consequences [10].
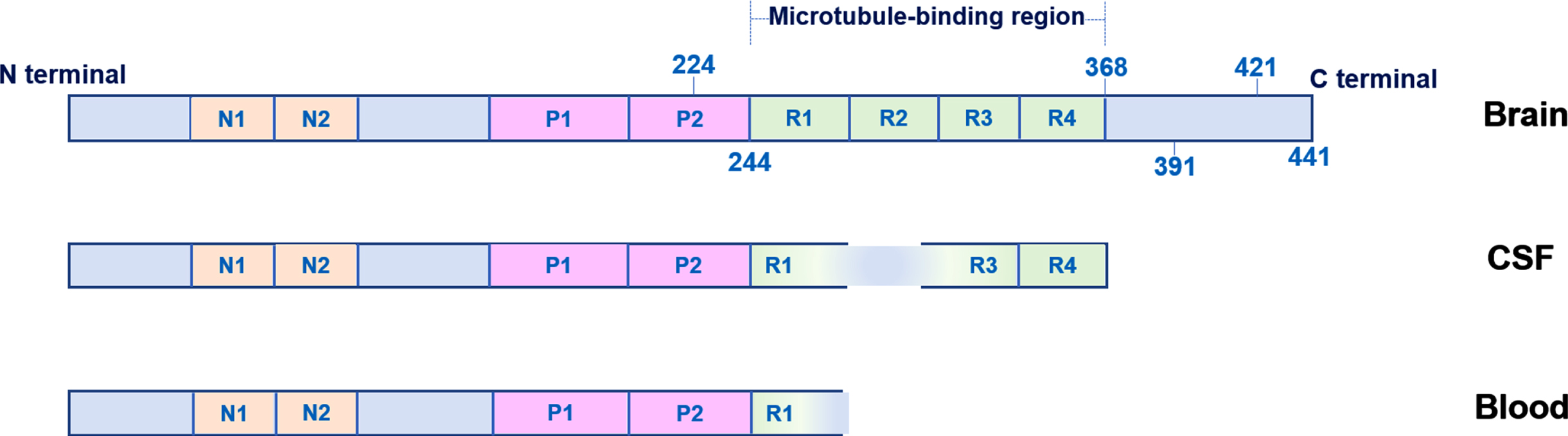
Tau protein in the brain, cerebrospinal fluid (CSF), and blood.
The entire tau protein in the brain is shown in Fig. 3 [43]. Tau protein is modified post-translationally at several sites, including truncations at amino acids 368 [44], 391 [45], and 421 [45], which promote the formation of fibrillar aggregates according to mass spectrometric studies [46, 47]. Truncated C-terminal fragments of tau are present in brain tangles, while the majority of the soluble form consists of N-terminal and mid-region forms [48]. Some of these forms are released into the blood and CSF [43, 49]. The middle picture in Fig. 3 depicts CSF tau, that lacks the protein’s extreme C-terminus but has mid-region epitopes. Blood tau, which stretches from the N-terminal to the beginning of the microtubule-binding domain, is depicted in the bottom image (around amino acid 254). P-tau levels in the blood are relatively tiny (up to 5%) compared to those in CSF in the same individual [50], indicating that p-tau originating from the brain enters the blood through the CSF. Even though phosphorylated tau serves established physiological purposes, such as maintaining microtubule integrity and assembly [51], phosphorylation over a certain threshold has pathological effects [52, 53].
Neuropathology and PET tests can identify the gradual accumulation of certain fractions of the brain’s phosphorylated tau pool into insoluble filamentous tangles during AD [46, 54]. Additionally, certain soluble p-tau fractions are released into the CSF in increasing amounts, where they may be found and measured to offer a detailed indication of the presence of a disease [54–56]. Several epitopes, including amino acids 181, 199, 202, 205, 217, 231, 235, and 396, can be hyperphosphorylated in pathological tau [54, 57–59].
When compared to cognitively normal controls [60–63] and patients with dementias other than AD [50, 61–68], the amounts of plasma tau phosphorylated at various sites (p-tau181, p-tau217, or p-tau231) are much higher, more than two-fold.
Elucidated by mass spectrometry methods, CSF p-tau217 demonstrates higher disparities between people with AD and controls than p-tau181 [61, 69]. According to a significant multicenter cohort investigation, plasma p-tau217 can be a useful biomarker for differentiating AD from other types of dementia. Its accuracy rate is 96%, which is comparable to the performance of recognized CSF or tau-PET biomarkers [62]. The correlation between plasma p-tau217 concentrations and the density of cortical tau pathology in AD but not in other tauopathies, such as FTD-tau, emphasizes plasma p-tau specificity for AD tau pathology. These findings were made using neuropathological ratings of cerebral tau-tangle pathology [62]. Palmqvist et al. reported that levels of plasma p-tau217 increase approximately two decades before the onset of moderate cognitive decline in individuals with autosomal dominant AD. This finding is in line with previous studies that have suggested that abnormal levels of plasma p-tau217 occur before tau-PET [70]. This observation shows that the biological mechanisms underlying the tau-PET signal may not be the same as the cause of variations in plasma p-tau. Direct comparative investigations employing immunochemical and mass spectrometry platforms revealed that p-tau217 and p-tau181 effectively differentiate between various modalities, including tau-PET negativity, Aβ-PET negativity, autopsy-confirmed AD, and frontotemporal lobar degeneration [61, 69–72]. However, when employing mass spectrometry techniques, p-tau217 was more adept at identifying Aβ-PET positives [61, 69].
Another new p-tau marker that shows promise is p-tau231. The level of p-tau231 in the cerebrospinal fluid was found to be one of the earliest indicators of AD pathology in the brain, related to the accumulation of Aβ protein. According to both Aβ-PET and tau Braak stages [64], p-tau231 may be particularly useful as an early pathology marker of AD even before symptoms appear. The advantage of p-tau231 over p-tau181 or p-tau217 for predicting amyloid or tau-PET was not replicated in a different investigation of a community-based population [73].
p-tau231 and p-tau217 can help with enrolling participants in early-stage clinical trials for AD. However, the selection of a plasma biomarker may vary depending on the specific objective of the clinical trial. For instance, p-tau231 might be the most appropriate option for trials involving middle-aged individuals who have alterations in soluble Aβ but not enough Aβ pathology to be detectable on PET scans [74]. Other plasma biomarkers may work well for older individuals or for trials in people with established Aβ pathology on PET scans [74].
Blood-based biomarkers
Numerous concepts about the pathophysiology of AD have led to the creation of multiple subtypes of biomarkers. Those biomarkers are linked to certain pathogenic mechanisms, including the deposition of Aβ, tau-associated neurofibrillary tangles, and neuroinflammation. When compared to CSF, the Aβ42/Aβ40 ratio in plasma is only reduced by 14–20% [75–78], in contrast to a 50% reduction in CSF [35] in AD sufferers. The weak correlation with CSF can be explained by the production of Aβ peptides in platelets and other non-brain tissues [9]. The discovery on AD biomarkers can be summarized in Table 1.
Some of the most used AD blood biomarkers
A desperate demand exists for diagnostic methods that are convenient, affordable, non-invasive, and readily accessible diagnostic techniques, such as blood testing, to identify the illness. Researchers are monitoring the levels of particular markers in the blood to see if they may accurately predict changes associated with AD. These biomarkers may be evaluated before and after symptoms manifest, including tau, Aβ, or other compounds. By assisting in identifying clinical trial patients and raising the prospects of early diagnosis and intervention, these testing technologies can aid in discovering new drugs. Additionally, a blood test would make it possible to evaluate the development of AD in more substantial, diversified populations [33].
Genetic risk profiling
Three genes with unusual variants have been found to cause AD, and numerous more genes have appeared to enhance AD possibility. The first gene revealed to have mutations causing a hereditary type of AD is APP. Scientists from all around the world are looking for genes that may reduce a person’s risk and new dangerous genes. With the introduction of more effective medicines, genetic profiling may turn into a useful risk assessment approach [33].
The brain’s primary lipid transporter, apolipoprotein E (ApoE), supports peripheral nerve damage brought on by AD and nerve regeneration. The ApoE receptor belongs to the family of low-density lipoprotein receptors, and it facilitates the transport of cholesterol to cells within the central nervous system, including neurons. The main source of ApoE is astrocytes, and there exist three main subtypes of ApoE: ApoE2, ApoE3, and ApoE4. One of the most significant genetic risk factors for sporadic AD is APOE4, which is closely related to the neuropathology induced by Aβ. Additionally, the existence of APOE4 exacerbates the harm to the brain caused by tau [83, 90]. Various clinical trials have employed APOE4 genetic testing to identify people at high risk for AD or at risk for related adverse effects to authorize therapies. APOE4 is the most vital risk gene in some populations.
Over the past ten years, genome wide association studies (GWAS) have identified various crucial genetic risk factors, including but not limited to, ABCA7, BIN1, CD2AP, CLU, CR1, EPHA1, MS4A6A-MS4A4E, and PICALM genes, in addition to the APOE4 allele variant [91]. GWAS has found that a certain area near the BIN1 gene is a major factor in causing AD [92]. The research has revealed that individuals with AD have elevated BIN1 gene expression levels in their brains, and that a specific genetic variation in the BIN1 gene, referred to as a 3bp insertion, regulates tau protein levels and raises the possibility of developing AD [92]. Bellenguez et al. confirmed that AD involves the amyloid and tau pathways and suggested that a certain type of immune cells called microglia may be involved. In addition, the research identified 31 previously unknown genes that could potentially be linked to AD and created a novel genetic risk assessment that can anticipate a person’s risk of developing the disorder [93].
CURRENT STRATEGIES OF AD DETECTION
Brain imaging
Neuroimaging, often known as brain imaging, is frequently utilized nowadays to aid in the diagnosis of AD. Research is still advancing, offering novel and sophisticated methods for imaging the brain [33]. Three types of neuroimaging methods are available: structural imaging techniques such as magnetic resonance imaging (MRI) and computerized tomography (CT), functional imaging techniques such as positron emission tomography (PET) and functional MRI (fMRI), and molecular imaging techniques such as PET, fMRI, and single-photon emission computed tomography (SPECT). Structural imaging allows for the examination of the shape, position, and volume of brain tissue. Alternatively, functional imaging reveals how effectively cells across various brain areas are performing their functions by indicating their use of glucose or oxygen. MRI is a non-invasive technique of analyzing depositions. Molecular imaging scans for cellular or chemical alterations linked to specific diseases using highly focused radiotracers [94, 95].
Positron emission tomography
Amyvid ([18F]florbetapir), Neuraceq ([18F]florbetaben), and Vizamyl ([18F]flutemetamol) are amyloid PET tracers that have been approved by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for use in assessing patients with cognitive impairment who may have AD [96]. A radiotracer, [11C] PiB (Pittsburg compound B), is often utilized in research. However, due to the brief half-life of carbon-11, which is around 20 minutes, it is necessary to have a cyclotron located on-site to manufacture it [9].
The PET tracers used for Aβ imaging are designed to selectively bind to amyloid deposits, allowing for visualization of the plaques in the brain. This specificity is achieved through the use of chemical modifications to the tracer molecules that enhance their binding to Aβ deposits, while reducing binding to other brain components. As a result, the images obtained by Aβ PET tracer offer a method to evaluate how amyloid pathology is distributed and advancing in the brain, which is crucial for the detection and treatment of AD [97].
Figure 4 depicts an example PET picture of amyloid. In its last 15 years of deployment in research, amyloid PET has overcome several challenges. Amyloid PET has been neuropathology verified [96], experienced substantial standardization about the quantification of Aβ pathology and the determination of cut-points for abnormalities [99] and has solidified adequate usage criteria [100]. When an anti-amyloid treatment is authorized, amyloid PET has emerged as a primary solution for clinical use and is currently one of the most frequently utilized biomarkers in clinical trials. The accessibility of PET imaging and cyclotron manufacturing facilities varies between countries and greatly influences the use of amyloid PET. The availability of PET imaging time and the manufacture of radiotracers for imaging and treatment monitoring of amyloid treatments are expected to have an impact on the wait times for treatment of patients, at least initially, until infrastructure upgrades can be implemented. According to the RAND Corporation, using CSF biomarkers as an alternative or in conjunction with amyloid PET may lessen the wait times for amyloid treatment in several countries, i.e., Japan [101], the US [102], Europe [103], and Canada [104].
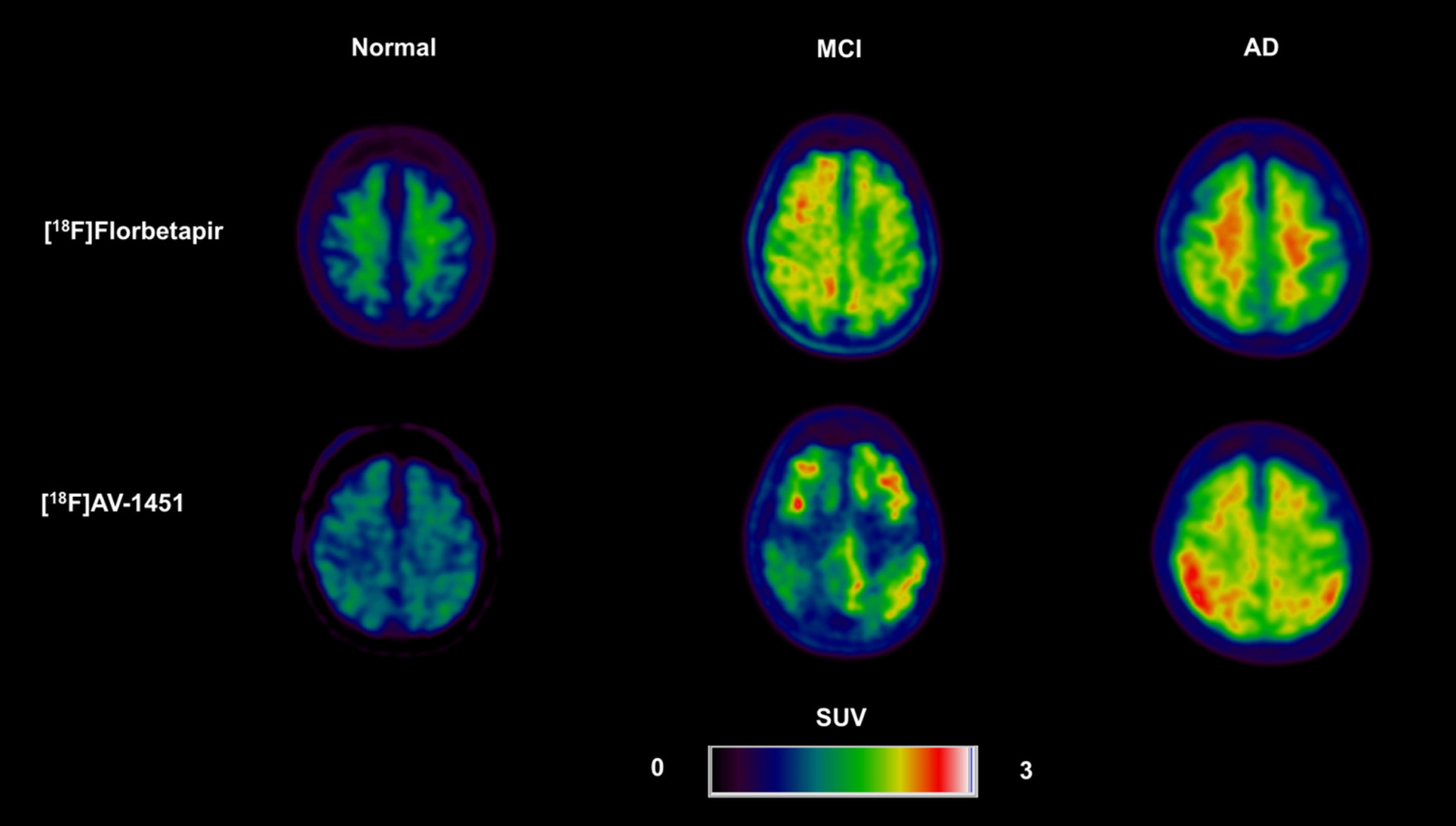
Amyloid PET between normal, mild cognitive impairment (MCI), and Alzheimer’s disease (AD) patient [98].
Compared to amyloid PET, tau imaging is still in its infancy concerning accessibility and standardized use due to several reasons: Firstly, there is a limited number of approved tau PET tracers, and they are not widely available in all the regions of the world. Secondly, the interpretation of tau PET images can be challenging and requires specialized expertise. Additionally, the relationship between the presence of tau proteins in the brain and the observable symptoms of AD is not yet clearly defined, which poses challenges to the clinical usefulness of tau PET imaging. In addition, the practical aspects of tau PET imaging, such as the uptake of tracers, may be impacted by age, metabolism, and genetics, which can make it challenging to achieve consistent findings across various investigations. Additionally, there is a shortage of comprehensive, meticulously executed clinical studies that have been done to evaluate the effectiveness and dependability of tau PET imaging in a real-life context. All of these factors contribute to the limited accessibility and standardized use of tau PET imaging compared to amyloid PET.
Additional advances are required before they may be used in a clinical context. There is still work to be done in areas such as developing standardized processing techniques, establishing cut-off points for positive results, and collecting more data. However, the results of PET imaging studies that have been conducted so far support the Braak model, which suggests that there are specific spatial and temporal stages in the progression of tau pathology in the brains of individuals with AD [46]. Clinical trials have demonstrated the potential of tau PET in detecting pharmacodynamic effects of disease-modifying drugs on Aβ (tracking tau changes further downstream) and tau pathology (target engagement) [105]. CSF testing, however, may be considered invasive. Meanwhile, PET imaging is an expensive modality with restricted accessibility, and involves minimal exposure to radiation.
Magnetic resonance image
MRI technology uses the magnetic fields of hydrogen atom nuclei to generate images [106]. Essentially, the oscillation of these nuclei may be controlled and used as tracers, which, in turn, provides a signal that can be used to construct a picture [107].
MRI technology employs radio waves and magnetic fields to create high-quality 2D and 3D pictures with high levels of contrast and resolution. This method is classified as noninvasive and nonharmful neuroimaging because it does not include the use of any radioactive tracers or hazardous radiation from X-rays. The most popular MRI techniques for diagnosing and classifying AD include structural MRI, which enables specialists to assess brain volumes in images and identify areas of the brain that have lost tissue, cells, or neurons.
Medical professionals frequently employ MRI as a radiation-free standard diagnostic method for illness diagnosis, staging, and follow-up. Almost every portion of the body can be examined with an MRI because every scan produces a precise, three-dimensional anatomical picture. Regarding AD, MRI technology enables a detailed 3D model of the subject’s brain to be created [108]. The ability to examine brain structures and their changes over time is a crucial function of MRI in the diagnosis of AD. This way, alterations in the hippocampus, frontal, and parietal areas indicate the disease’s progression to dementia [109].
The movement of molecules in tissues can be different in different directions due to their physical arrangement or obstacles. MRI uses magnetic field pulses to measure this movement, but only in the direction of the pulses. By changing the direction of the pulses using diffusion technique, the movement in different directions can be seen, making diffusion measurement a unique and powerful tool in MRI. This technique is called diffusion tensor MRI (DT-MRI), a type of brain imaging that looks at the movement of water molecules to understand the structure of the white matter in the brain. It measures how easily water molecules can move around structures like white matter fibers or cell bodies. In white matter, the speed of water movement is affected by things like the size of axons and how much myelin is present [110].
DT-MRI uses several measurements to understand the movement of water in tissues. One of these is the fractional anisotropy (FA), which measures how much the speed of water movement changes depending on the direction it’s measured in. If the water moves at the same speed in all directions, the score is 0, and if it moves in only one direction, the score is 1. Another measurement is the mean diffusivity (MD) that shows the average speed of water movement. Other measurements like the radial diffusivity (RD) and longitudinal diffusivity (LD) show the fastest and slowest speeds of water movement. All of these measurements help researchers understand how changes in water movement are related to changes in the structure of white matter. Harrison et al. suggests a connection between AD risk and alterations in the movement of water within the white matter of the brain [111]. Several white matter tracts showed decreased FA and increased MD, RD, and LD. It is essential to detect and treat AD early, as DT-MRI is advancing and may eventually furnish useful markers and information for the condition [111].
Nanoparticles detection
Magnetic iron oxide nanoparticles
Iron oxides can be used as MRI contrast agents or drug carriers since they are harmless, biocompatible, and can be injected into humans [112]. They can also be absorbed into the patient’s normal metabolic processes. It is possible to modify iron oxide nanoparticles (IONPs) to exhibit traits such as solubility and biocompatibility, and their tiny size allows them to access brain regions that are otherwise inaccessible. By enclosing the IONPs with different substances, multiple iron nanoparticle formulations have been produced that serve as magnetic resonance contrast agents. Studies on mice with neuroinflammation have shown that intravenously administered IONPs can penetrate the brain via the choroid plexus [113].
Magnetic nanoparticles (MNPs) tagged with antibodies have been proven to identify Aβ and tau protein in blood, CSF, and serum-like samples for the in vitro diagnosis of AD [114–116]. Magnetic IONPs coated with the anti-biofouling polymer polyethylene glycol-block-allyl glycidyl ether (PEG-b-AGE) could detect Aβ40 and Aβ42 peptides and tau protein in CSF and serum-like samples with a sensitivity of over 95% and specificity of over 90% when corresponding antibodies were used as targeting ligands [114]. Additionally, the sensitivity of antibody-conjugated anti-biofouling coated IONPs for detecting tau and Aβs protein in blood samples obtained from humans was greater than that of antibody-conjugated superparamagnetic iron oxide nanoparticles (SPIONs) [114]. Comparing the anti-biofouling performance of IONPs and SPIONs, it was found that IONPs did not exhibit a substantial decrease in sensitivity for detecting tau protein or Aβ40 in artificial CSF and fetal bovine serum (FBS)-supplemented phosphate-buffered saline (PBS) (with a range of 78.2 to 100% sensitivity in CSF-like medium and 83.9 to 97.8% sensitivity in FBS-added PBS). On the other hand, SPIONs showed a decrease of approximately 40–60% (from a range of 82.3–100% sensitivity in CSF-like medium to 43.1–56.4% sensitivity in FBS-added PBS) [114]. Jiang et al. satisfactorily measured the amount of Aβ oligomer in synthetic CSF by using Fe3O4 magnetic IONPs as detection and concentration agents with an aptamer that targets Aβ oligomers and a complementary oligonucleotide of the Aβ oligomer. Fluorescent labeling was accomplished using BaYF5:Yb, Er upconversion nanoparticles [115].
For the AD in vivo diagnostic, SPIONs conjugated with anti-AβPP) antibodies can be used to picture the amount of plaques in APP/PS1 transgenic mice [117]. The APP/PS1 transgenic mouse model is a specific type of mouse that has been genetically modified to contain mutations in the genes APP and PSEN1, which are associated with familial AD (FAD), an early onset form of AD. These mutations result in the excessive production of Aβ, which is believed to be a key factor in the development of AD. The APP/PS1 mouse model is significant to the current discussion because it is an effective resource for investigating the mechanisms involved in AD, and for evaluating potential treatments for the disease. By observing the effects of the FAD mutations in a living organism, researchers can gain valuable insights into the disease process and explore new approaches for treating AD.
The brains of NMRI mice can have amyloid plaques marked by ultrasmall SPIONs that are linked to the peptide C-IPLPFYN-C, also known as USPIO-PHO [118]. Amyloid deposits within the brains of Tg2576 mice may be found using SPIONs coupled to curcumin [119]. Fibrin γ377–395 peptide-coated Fe2O3 nanoparticles can specifically inhibit microglial cells in rTg4510 mice that carry the tau mutation, offering a potential therapeutic approach against neurodegenerative tauopathies [120].
In recent work by Fernandez et al., magnetic IONPs conjugated to an antiferritin antibody to identify ferritin protein in specific brain areas of a mouse model that has been genetically modified. The mice used were the hemizygous strain 5xFAD, which contained five mutations linked to familial AD. The transgenic mice carrying mutations in APP, including Florida (I716V), Swedish (K670N, M671L), and London (V717I) mutations, along with L286V and M146L mutations in presenilin 1 (PS1), show certain characteristics of AD observed in humans, such as the occurrence of amyloid plaques. Through the use of functionalized IONPs, the ferritin protein that accumulates in the subiculum region of the AD-afflicted mice can be specifically identified and bound to [121].
Quantum dot detection
Fluorescent semiconductor nanocrystals, often called quantum dots (QD), can be a valuable resource in constructing a sensitive lateral flow assay (LFA) because of their remarkable luminosity, resistance to photobleaching, stability against chemical and thermal influences, and simplicity of surface customization [122]. QDs have a core size that ranges from 1 to 10 nanometers. To use them in biological applications, their surfaces are usually modified with a bio-functional layer which makes the QDs about 10 to 30 nanometers in size. This surface functionalization helps with the dispersion of QDs in water, as well as affecting their electrochemical characteristics and their ability to interact with biomolecules.
QDs are increasingly used as tracing agents for several neurological illnesses. Due to their distinctive optical characteristics, QDs have advantages in nanomedicine: at a nanoscale range, they provide remarkable stability, sensitivity, and selectivity, thus rendering them a promising theragnostic tool for AD [123]. However, their toxicity has also been a concern in recent years. Evidence suggests that QDs can be toxic to cells and cause oxidative stress, leading to cellular damage and death [124, 125]. In addition, when ingested or inhaled, they may also be toxic to various tissues and organs, including the liver, lung, and heart [126]. Moreover, the toxicity of QDs can vary depending on factors such as size, composition, surface coating, and exposure route [127].
By utilizing the lab-on-a-chip technology, it is possible to examine the electrochemical characteristics of QDs as markers and apply them in detecting the ApoE biomarker with exceptional precision and sensitivity. The method achieves a limit of detection (LOD) as low as 12.5 ng/mL and a linear range of 10 to 200 ng/mL, exhibiting excellent accuracy for diluted human plasma samples. This approach allows for an assessment of ApoE as a potential biomarker for diagnosing AD [128].
Feng et al. aimed to address the absence of effective diagnostic methods for AD, a QD probe was conjugated with an anti-Aβ antibody (QD-Aβ-Ab) [123, 129]. After being injected intracerebroventricularly into both normal mice and mice carrying mutant human APP695swe and APP717 V-F transgenes, this QD-Aβ-Ab was then photographed. The results showed that Aβ42 was present in the CA1 region of the hippocampus in the APP transgenic mice. Fluorescence microscopy showed that the fluorescence was mainly found in the hippocampus, sagittal septum, striatum, and cerebral cortex of the APP transgenic mice. Healthy mice displayed a narrower range of fluorescence and a lower fluorescence signal compared to the APP transgenic mice, according to in vivo imaging of mice given the QD-Aβ-Ab probe. As comparison to the 10- and 16-month-old APP transgenic mice, the average fluorescence signal in the brain tissues of healthy C57BL mice was much lower. This work shows that QD-Aβ-Ab can monitor Aβ accumulation in vivo and raises the possibility that these probes might be employed for initial molecular imaging of AD. For sensitive and accurate sandwich immunoassays that detect Aβ42, QDs have also been employed as fluorescent labels [129].
Tang et al. coupled Streptavidin-QDs to biotinylated anti-Aβ 1-16 (N-Ab) [130]. The C-terminal antibody and the biotinylated N-terminal antibody can specifically attach to Aβ42 to generate sandwich-like immunocomplexes, C-Ab-Aβ42-N-Ab. As a result, the fluorescence intensity of the supernatant can be used to measure the quantity of Aβ42, making it a notable and quick approach for AD detection, for which the LOD is 7.6 pg/mL [130].
Carbon QDs and graphene QDs are two varieties of carbon core-type QDs [131]. Graphene QDs inhibit the development of amyloid plaques and prohibit the cytotoxicity brought on by Aβ oligomers, a result due to their ability to pass the blood-brain barrier and their low cytotoxic nature. The ability of Aβ42 peptides to attach to carbon materials in a hydrophobic manner decreases the negative surface charge and enhances the inhibition effect of QDs [132]. Selected applications of QDs are summarized in Table 2.
Quantum dot applications in neurodegenerative disorders
GQDs, graphene quantum dots; CQDs, carbon quantum dots.
Gold nanoparticle detection
Gold nanoparticles (AuNPs) are a popular choice for labeling in LFA. They are spherical, inert, and have a strong attraction to biomolecules. Their size and optical properties can be adjusted, making them a versatile tool for LFA. Furthermore, they are environmentally friendly, stable, and provide a strong optical signal for analysis. This signal can be further amplified with the addition of other substances like silver nanoparticles [138].
In neurodegenerative illnesses, AuNPs can bind valuable compounds and improve drug transport over the blood-brain barrier. Additionally, AuNPs exhibit unique optical features, nanoscale chirality, antioxidant impact, and anti-amyloidosis activity. Due to their remarkable qualities, they have received a lot of attention in the research on AD diagnosis [139].
AuNPs are highly valued in the fields of medicine and biology, as they have the possibility to be utilized in treating neurodegenerative diseases. Their ability to work in unison to suppress Aβ aggregation, dissociate Aβ fibrils, and prevent deficits in spatial and recognition memory is particularly noteworthy [131]. Moreover, AuNPs also have exceptional optical qualities [140], as well as catalytic activity, electrical conductivity, chemical stability, and biocompatibility [140–142]. With their large surface area and strong protein adsorption abilities, they can effectively bind with Aβ proteins when combined with antibodies. Furthermore, they improve the sensor signal and enhance the effectiveness of electron transfer [16].
Consequently, AuNPs have been extensively utilized to create biosensors for identifying AD biomarkers [139]. Still, a few toxicity issues exist with AuNPs. Aβ42-induced cytotoxicity cannot be successfully prevented by the bare AuNPs [143]. More peptide-functionalized AuNPs are being investigated for the purpose of reducing the cytotoxicity caused by Aβ42. The selected AuNPs-associated sensors for the detection of AD are summarized in Table 3.
In vitro Alzheimer’s disease detection using Au nanostructures-related sensors
Nanodiamond detection
Nanodiamonds (NDs) have the sp3 tetrahedral monocrystalline carbon nanoparticles as the core [152]. Some nanodiamonds may have a sp3 carbon coating, which is made up of pure carbon, while others may have a sp2 coating, made up of a mixture of carbon and nitrogen atoms. Additionally, some nanodiamonds may be functionalized with various chemical groups, such as carboxyl or amine groups, which can alter their surface properties and allow for specific interactions with biological or other materials [153].
NDs with sizes ranging from 2 to 100 nm are biocompatible and may be functionalized to deliver therapeutic molecules to particular sites. Also, NDs can bind to multiple drugs. Negatively charged nitrogen-vacancy (NV–) centers in a high-density ensemble with exceptional optical and magnetic properties is also present in ND. To accomplish background-free detection in tissue imaging, either a sinusoidal magnetic field or microwave radiation can be used to regulate the fluorescence intensity of NV–.
Remarkably, ultra-nanocrystalline diamond films have been proven to favor neural stem cell differentiation [155, 156]. Alawdi et al. proved that by reducing NMDA (Glutamate) receptor stimulation and avoiding nerve injury, ND could have a neuroprotective effect to reverse symptoms induced by aluminum oral intake, which resemble AD. The inflammatory cytokines IL-6 and TNF-α decrease when the brain-derived neurotrophic factor increases. NDs increase the phosphorylated STAT3 and reduce the level of NF-kβ (nuclear factor kappa β), preventing neuronal death. Additionally, the NDs successfully accelerated the clearance of p-tau protein and Aβ plaques [157].
Moralez-Zavala et al. announced the functionalization of steady fluorescent markers based on nanodiamonds with a bifunctional peptide [158]. According to their findings, functionalized NDs (fNDs) may be internalized by cells and are not cytotoxic. The fNDs enable susceptible detection of in vitro Aβ fibrils and Aβ aggregates by labelling ex-vivo brain slices of AD mice (at picomolar concentrations of NDs). In comparison to frequently used fluorescent markers (such as Thioflavin T, which is used to stain Aβ aggregates), the fNDs provide higher steady fluorescence. Unfortunately, there is a lack of control sample in this study, which limits the reliability of these findings in detecting Aβ aggregates crucial to AD pathogenesis [158, 159].
Polymer detection
Numerous conducting polymers have been employed in biosensing purposes, including polypyrrole (PPY), which has drawn much experimental attention because of its excellent thermal/environmental stability, simplicity in manufacturing, biocompatibility, and programmable electrical characteristics. Supraja et al. studied a portable label-free chemiresistive sensing device based on PPY nanoparticles to concurrently detect Aβ40 and Aβ42 peptides biomarkers using spiked-PBS and spiked plasma specimens. By performing this, they could create an array from easily accessible, inexpensive copper clads, enabling the detection of both biomarkers on a single platform. This multianalyte sensing capability on a single platform is a significant advancement since in a single step, the target peptide ratio in a human sample may be determined [160].
Polyvalent directed peptide polymer (PDPP), created by Lee et al., is a multivalent peptide probe that increases binding sensitivity and specificity by cooperating with the target molecule [161]. Lee et al. used a fluorescence detection technology based on nanoporous zinc oxide (ZnO) for AD detection at the presymptomatic period, creating a unique ultrasensitive diagnostic device. To improve binding affinity, sensitivity, and selectivity, they bound several target sites. In comparison to a single FITC-labeled peptide, the polyvalent exposure of PDPP to Aβ42 increased sensitivity by around 104-fold. The peptides in PDPP form demonstrated significantly greater affinity to Aβ42 compared to individual peptides. Given its large specific surface area, nanoporous ZnO demonstrated effective amplification of fluorescence. Additionally, the probe’s effectiveness as a powerful initial-stage AD detection method was evaluated using CSF samples from AD patients. The LOD for Aβ42 was dramatically decreased by the binding of PDPP to the nanoporous ZnO structure, going from 100 pg/mL for the usual antibody-based method normally used in health center [162] to 12 ag/mL [161].
Due to their magnetic characteristics, magnetic beads (MBs) have been widely utilized as sensors in biosensing. MB is a polymer sphere made up of nanomagnetic particles that are randomly distributed [163]; as a result, the material is still superparamagnetic but responds quickly to magnetic fields outside of it [164]. They make purification and separation easier by precondense the specimen and by minimizing matrix outcomes, thereby making it easier to analyze actual samples.
Various type of polymers, namely poly(lactic-co-glycolic) acid (PLGA) [165–167], can be utilized in the production of MBs. PLGA is a type of polymer that is both biodegradable and biocompatible. As it has been accepted from both the FDA and the EMA, PLGA is frequently used to encapsulate various chemicals for diagnosis in biomedicine [168]. Tau protein was detected using MB@NAV in a sandwich immunoassay technique. The LOD was 63 ng/mL [169].
Optical analytic platforms
Enzyme-linked immunosorbent assay (ELISA)
Since peptides and tangles are the primary biomarkers of AD, an ELISA is frequently employed to measure them. The conformation specificity of the ELISA-used antibodies significantly impacts this approach. ELISA is known for its excellent specificity, sensitivity, and low dependency on sample preparation [170]. In ELISA, a measurable signal that identifies substances, like a change in absorbance, is produced by the binding interaction between a ligand and an antibody. The detection range of ELISAs used in fundamental research and commercially accessible ELISA kits is often in the picomolar range (pM) [171].
Numerous researchers initially created direct ELISAs, which include coating microwells with brain homogenate or pure paired helical filament, which are subsequently detected by anti-tau antibodies [172–174]. High background signals resulting from nonspecific sample binding to the plate can cause direct ELISAs to exaggerate or understate the concentration of target proteins, even when standard proteins are employed. This is the main drawback of direct ELISA [173, 174]. Several groups have also constructed sandwich ELISAs to find phosphorylated tau levels in the brain. Yet, they only employed buffer-soluble brain tissue (i.e., that free of detergent), which could dramatically underestimate the quantity of insoluble tau buildup [175].
Sandwich ELISA has the advantage of quantitating bio-samples. Sandwich immunocomplexes, which are produced by antibody pairs that can successfully trap the target analyte into the well, are used to measure protein concentration. Thus, the immunocomplexes are comprised of capture antibody-analyte-detection antibody. An enzyme that catalyzes the transformation of the substrate to the product is attached to the detection antibody, causing it to fluoresce or change color correspondingly to the analyte concentration in the specimen [176].
According to a recent study, the plasma Aβ42/Aβ40 ratio, as measured by ELISA, accurately predicts the cerebral Aβ pathology compared to the Aβ42/Aβ40 ratio of amyloid PET [75–77, 178]. Figure 5 shows a scheme about digital ELISA [177], where the left panel shows how MBs with antibodies attached are mixed with a target protein to create protein-bead complexes. The complexes are then labeled with another antibody that has an enzyme attached. The right panel shows two examples, one with a low amount of protein and one with a high amount. Next, the protein-bead complexes are placed in a microwell array with tiny wells, each capable of holding one bead. A substrate is later added that fluoresces when it reacts with the enzyme. By counting the number of beads with a protein molecule attached, images of a tiny portion of the well array are captured and the fluorescence is utilized to determine the concentration of protein in the bulk solution [177].
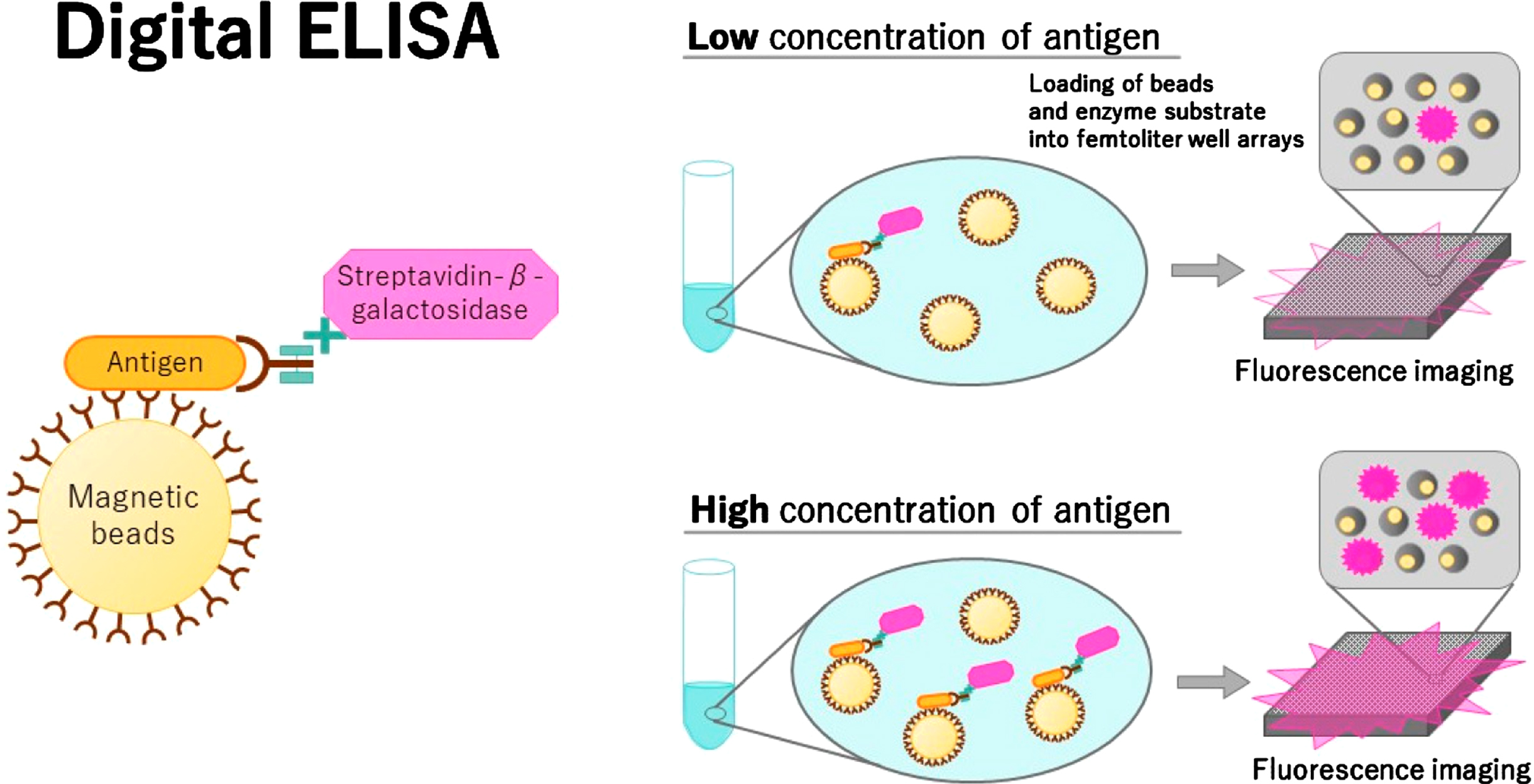
Digital ELISA [177].
Perez-Ruiz et al. established a magnetic particle-based digital ELISA for the attomolar measurement of the tau protein in the CSF samples and plasma at ultrasensitive levels. A LOD of ∼55 aM and ∼24 and in plasma and buffer and samples, respectively, was achieved using the chosen high-affinity antibodies in conjunction with the most suitable test circumstances. Compared to commercial ELISA (Total Tau ELISA, Euroimmun, Germany) with a LOD of ≤0.2 pM, this digital ELISA was around 5000 times more sensitive [179].
As a typical sensing technique, ELISA has been widely used to detect amyloid oligomers in bodily fluids. However, ELISA application is less preferrable due to the cross-reactivity and nonspecific binding. This takes a lot of time and is expensive. Moreover, the immunosorbent assay performance is susceptible to the CSF’s salinity environment, limiting its usefulness [180–182]. Hence, finding biomarkers of AD in biological samples that can be detected with high sensitivity and selectivity is still a difficult task.
Surface-enhanced Raman scattering (SERS)
Recently, there has been a rise in the use of Raman and SERS for disease detection. The reasons for their increased application have frequently been attributed to their well-known superiority, which includes the generation of tapered spectral bands as a feature of the molecular compounds contained, their non-destructive analysis method, and the sensitivity and specificity they are harnessed with.
SERS is a potential method for quickly identifying small concentrations of target compounds. It amplifies Raman signals up to a 104–107 magnitude, outperforming Raman spectroscopy [25]. SERS is now a potent strategy for monitoring selective recognition due to its extremely high specificity and sensitivity, especially in biosensing, surface research, and environmental analysis [183]. For the time being, plasmonic materials, such as gold and silver, are frequently employed as SERS substrates for SERS-based biological detection [20, 185]. Label-free SERS has established itself as a potent method for investigating protein kinetics and structure. Of course, the use of a suitable substrate is essential for SERS. As a result, a lot of work has been put into developing metallic nanostructures that are SERS-active for protein identification.
The recognition of t-tau proteins and Aβ peptides has been achieved using SERS, a vibrational spectroscopy approach [23, 186]. This creates a precise molecular fingerprint that enables quick recognition of target biomarkers. Using t-tau and Aβ42 peptide protein’s SERS spectra following their specific binding to the appropriate antibodies, it is possible to determine the details of the analytes’ chemical bonds, such as the protein-CN band (Raman peak at 1090 cm–1), histidine band (1,440 cm–1), and amide I band (1,650 cm–1). The outcomes showed that the SERS-based label-free sensing device could detect Aβ42 peptide at 500 fg/mL and t-tau protein at 100 fg/mL [187]. SERS-based tests from clinically relevant specimens have not been examined. A silver nanogap shell (AgNGS) SERS colloidal nanoprobe was created and produced by Yang et al. for the accurate and sensitive measurement of Aβ40 and Aβ42 in blood [188]. Ag-thiolate lamella structures formed by Raman-tagged chemicals for SERS signal generation regulate the nanogap at 1 nm resolution by altering the reaction rate for AgNGS synthesis (Fig. 6). The minimum detectable biomarker concentration is 0.25 pg/mL.
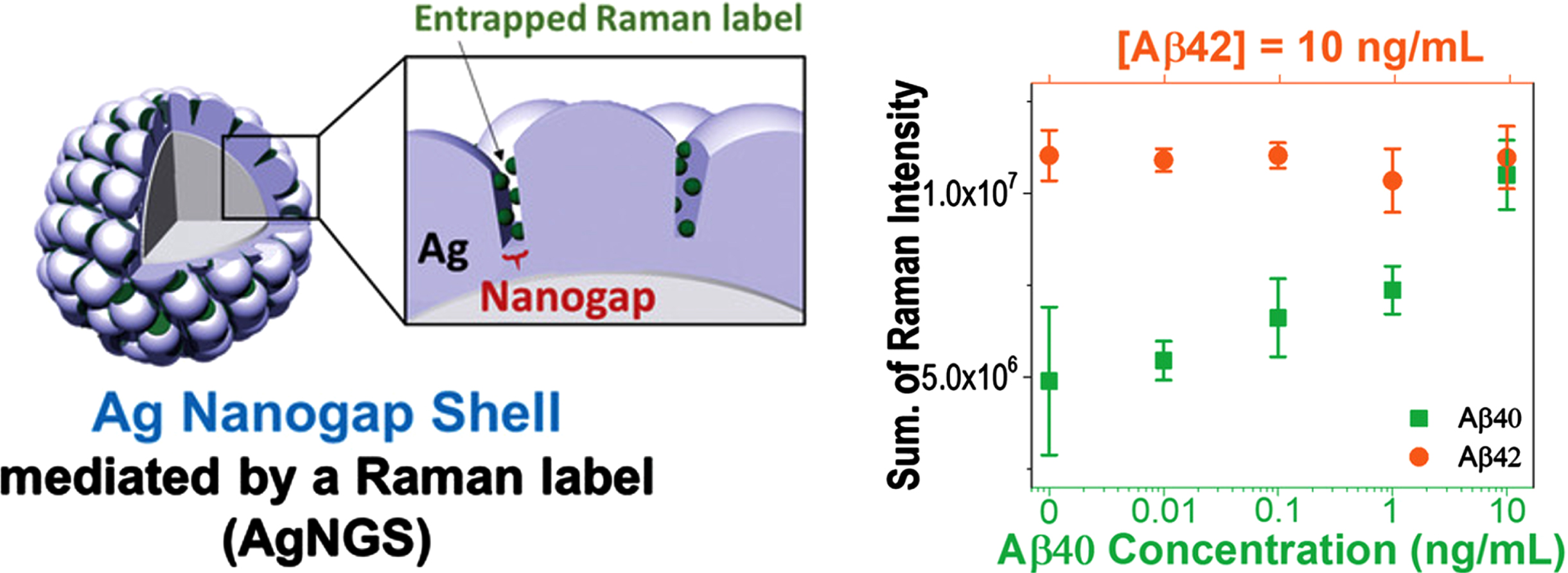
Schematic diagram for the Raman-mediated production of AgNGS and assessment of the cross-reactivity of AgNGS nanoprobes targeted for Aβ40 or Aβ42 [188].
Graphene sheets enhanced with magnetic nanoparticles have shown upper capability in biological area, surpassing that of pure GO and pure Fe3O4 [187, 189]. Yu et al. created a SERS-based immunoassay using particular antibodies linked to Fe3O4@GOs and 4-mercaptobenzoic acid (4-MBA) labeled silver probes [20]. The target protein has been captured, as seen by the 4-MBA SERS spectrum. Its ability to identify biomarkers at the fM level in clinical serum samples opens new avenues for their identification [20]. Developing sandwich ELISA platforms based on SERS and using SERS to identify biomarkers in clinical CSF or blood specimens may transform SERS into a real AD diagnostic tool. Selected applications of SERS and ELISA for AD detection are summarized in Table 4.
SERS and ELISA application in AD detection
CONCLUSION AND FUTURE OUTLOOKS
AD diagnosis is frequently established after irreparable brain damage has already occurred. With this in mind, creating techniques to identify AD biomarkers with affordable prices, noninvasively, portability, and time-efficiency is the principal mission for discovering this illness. Over the years, numerous conventional and modern techniques have been employed for detecting AD. For instance, detection of biomarkers in lumbar puncture samples and brain imaging are the two most common traditional techniques. In this study, we have summarized publications over the last few years that have dealt with the advancement of research in the field of early AD detection, which also employs several nanoparticles to detect several AD biomarkers, the most popular of which are the Aβ and the tau protein.
These developments are advantageous for research since they enable a more comprehensive range of complicated disorders with challenging diagnoses, including diseases like AD. Additionally, this information has helped develop diagnostic methods that are less dangerous than those already in use. Thus, the novel nanomaterial-based detection will make it possible to shorten the time required for imaging, enhance the stockpiling of relevant information, and, most importantly, enable an early diagnosis, which is required for improving the quality of life for AD patients.
The cost and availability of nanodiamonds, gold nanoparticles, magnetic oxide iron nanoparticles, QDs, ELISA, and SERS for AD diagnosis can vary depending on the specific application and the source of the materials. However, these technologies have shown promising potential for improving the accuracy and sensitivity of AD diagnosis. In terms of cost-effectiveness and availability, some of these technologies are more established and commercially available, such as ELISA, which is commonly used for measuring biomarker levels in blood samples. On the other hand, newer technologies like SERS and QDs are still being developed and optimized for clinical use, and their cost and availability may vary.
Nanodiamonds and gold nanoparticles have shown promise for use in portable platforms due to their biocompatibility and stability, while magnetic oxide iron nanoparticles have been used in portable MRI systems for AD diagnosis. QDs and SERS are still in the early stages of development, but they have shown potential for high sensitivity and specificity in AD diagnosis.
Thanks to the high sensitivity of photon detection-based immunoassays, ELISA and SERS have been investigated for AD detection in clinical samples. From the point of view of sensitivity, specificity, human applicability, and practicality, portable optical sensing approaches, like SERS and ELISA, offer a potential solution. ELISA and SERS have their own advantages and limitations. ELISA is widely used for its high sensitivity and specificity, as well as its ability to measure a wide range of analytes in a single assay. There is a reaction between antibody-antigen that makes both high specificity and sensitivity, and high efficiency because the analysis allows high throughput, where the test can be carried out simultaneously for thousands of samples. On the other hand, SERS is a highly sensitive spectroscopic technique that can detect trace amounts of biomolecules with high precision, making it a promising tool for early diagnosis of diseases like AD. However, SERS is limited by its dependence on surface enhancement and the presence of metallic substrates.
While both ELISA and SERS show promise for the early detection of pre-symptomatic AD, the adoption of these technologies in the clinical setting can be time-consuming and complex, so the adoption of these technologies in the clinic will likely require further validation through large-scale clinical trials. ELISA and SERS are two of the most advanced technologies for the clinical assessment of pre-symptomatic AD, and both hold promise for the early detection and diagnosis of AD. However, multidisciplinary research, further development and validation of these technologies will be necessary before they can be widely adopted in the clinic. For instance, although ELISA has been proven useful for the detection of elastin-derived peptides with an elevated concentration from CSF in stroke patients, it is not capable of measuring blood proteins of low concentration in CSF, including Aβ and tau proteins [204, 205]. Thus, the choice between ELISA and SERS for clinical assessment will depend on the specific requirements and goals of the study.
Footnotes
ACKNOWLEDGMENTS
This work was supported by National Science and Technology Council (NSTC), Taiwan, R.O.C.
FUNDING
Grant number: NSTC 109-2923-E-011-003-MY3, NSTC 111-2628-E-011-002-MY2.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.