
Abstract
Vitamin D is a secosteroid hormone exerting neurosteroid-like properties. Its well-known nuclear hormone receptor, and recently proposed as a mitochondrial transcription factor, vitamin D receptor, acts for its primary functions. The second receptor is an endoplasmic reticulum protein, protein disulfide isomerase A3 (PDIA3), suggested to act as a rapid response. Vitamin D has effects on various systems, particularly through calcium metabolism. Among them, the nervous system has an important place in the context of our subject. Recent studies have shown that vitamin D and its receptors have numerous effects on the nervous system. Neurodegeneration is a long-term process. Throughout a human life span, so is vitamin D deficiency. Our previous studies and others have suggested that the out-come of long-term vitamin D deficiency (hypovitaminosis D or inefficient utilization of vitamin D), may lead neurons to be vulnerable to aging and neurodegeneration. We suggest that keeping vitamin D levels at adequate levels at all stages of life, considering new approaches such as agonists that can activate vitamin D receptors, and utilizing other derivatives produced in the synthesis process with UVB are crucial when considering vitamin D-based intervention studies. Given most aspects of vitamin D, this review outlines how vitamin D and its receptors work and are involved in neurodegeneration, emphasizing Alzheimer’s disease.
INTRODUCTION
Vitamin D is a secosteroid (a steroid that has a “broken” B-ring) hormone exerting neurosteroid-like properties and is a primary ligand of vitamin D receptor (VDR) [1]. Calcium metabolism and bone mineralization are two well-known aspects of vitamin D. However, recent developments in vitamin D research suggest that further studies should be conducted on its lesser-known roles, which include immune system modulation, roles in cardiovascular disease, cancer, and regulation of neuromuscular and cognitive functions [2–4]. Our previous studies and others have suggested that the outcome of long-term vitamin D deficiency (hypovitaminosis D or inefficient utilization of vitamin D), may lead neurons to be vulnerable to aging and neurodegeneration [5–8]. In addition, we have proposed Alzheimer’s disease (AD) as a result of long-term hormonal-vitamin D imbalance [8–12]. We have already reviewed how mechanisms of vitamin D action and AD overlap in our previous articles [8, 11–13]. We discuss mechanism of vitamin D action and its receptors in neurodegeneration, focusing on AD.
THE DEFINITION OF THE MOLECULE, ITS SYNTHESIS, AND ORIGIN
What is vitamin D?
Today, vitamin D is considered one of the most critical factors for having and maintaining a healthy life [8, 15]. 1–25 dihydroxyvitamin D3 or 1–25 dihydroxycholecalciferol (1,25(OH)2D3) is a secosteroid hormone, the hormonally active form of vitamin D, and has neurosteroid-like functions. However, it is still classified as a vitamin due to its misnomer [8]. Certain enzymes produce this particular secosteroid hormone in the body of vertebrates, yet it was called a vitamin since when this hormone was first found, it was not known that it was produced in the human body [16]. This hormone shows its effects mainly, via the VDR, a nuclear hormone receptor [17, 18]. However, vitamin D derivatives and additional receptors will also be discussed later.
The synthesis or phototransformation of vitamin D and its metabolism
Vitamin D3 (cholecalciferol) is phototransformed by UVB from 7-dehydrocholesterol (7-DHC) in the skin, given that the skin continuously synthesizes vitamin D3 and releases it to the blood [19]. On the other hand, vitamin D2 (ergocalciferol) which can be regarded as a dietary intake, is also phototransformed by UVB by bond breaking of a bond on the fungal sterol ergosterol. The difference between vitamin D2 and vitamin D3 is the double bonds between C22 and C23 and a methyl group in the C24. Although both forms are accepted as equivalent, the diversity of the side chains, thus the hydroxylation sites, results in a lower affinity of vitamin D2 for 25-hydroxylase, VDR, and vitamin D binding protein[20].
Active vitamin D can be formed by the 25-hydroxylation in the liver and 1α-hydroxylation mainly in the kidney by cytochrome (CYP) enzymes. A set of 25-hydroxylases: mitochondrial enzyme CYP27A1, and the microsomal CYP2R1, CYP2J2/3, CYP3A4, CYP2D25, and CYP2C11 metabolize vitamin D to 25(OH)D3 [21, 22]. After 1α-hydroxylation by CYP27B1, 25(OH)D3 is converted to the active and circulating form of vitamin D the 1,25(OH)2D3 [23]. The CYP27B1 is strictly modulated by parathyroid hormone (PTH), fibroblast growth factor 23 (FGF23), and 1,25(OH)2D3 itself. Suppression of PTH by elevated calcium suppresses CYP27B1. The same result can be achieved by stimulating FGF23 through elevated phosphate. Vitamin D can also regulate its levels by increasing FGF23 production and inducing the catalytic enzyme CYP24A1 and inhibition of PTH [24–26]. FGF23 action requires membrane Klotho-FGF receptor complex. Mice lacking klotho or FGF23 exerts a premature aging phenotype [27].
1,25-hydroxyvitamin D3 24-hydroxylase (24(OH)ase), cytochrome P450 family 24, subfamily A, is an enzyme belonging to the class of polypeptide 1 (CYP24A1) and is the main regulator of 1,25(OH)2D3 level in tissues. It stimulates the catabolism of 25(OH)2D3 to 1,24,25(OH)3D3; therefore, it is an indicator of vitamin D utilization [28, 29]. Our studies showed the expression of this enzyme in cortical and hippocampal neurons [30], although 23-hydroxylase activity of CYP24A1 also results in biologically active 1,25(OH)2D3-26,23-lactone [31, 32].
Vitamin D3 can be isomerized to tachysterol3 (T3) and lumisterol3 (L3) by UVB in the plasma membrane of the keratinocyte [33]. 7-DHC, ergosterol, L3, and vitamin D3 and D2 were shown to be the substrates for CYP11A1 to produce several metabolites of vitamin D [26, 35].
The precursor of vitamin D, 7-DHC is also a precursor in cholesterol synthesis pathway [36–38]. The 24-dehydrocholesterol reductase (DHCR24) converts desmosterol to cholesterol [39]. Our study demonstrated that vitamin D effects DHCR24 levels by protein disulfide isomerase A3 (PDIA3) [40], suggesting the regulation of cholesterol synthesis pathway via vitamin D and PDIA3, in addition to VDR which downregulates the activity of sterol regulatory element-binding protein 2 (SREBP2) [40]. DHCR7 is functionally related to DHCR24 and suggested to regulate the cholesterol synthesis pathway. DHCR7 involves in the Kandutsch–Rusell pathway, where 7-DHC is converted to cholesterol [40–42]. Cholesterol enhanced loss of DHCR7 function associated with elevated vitamin D3 synthesis, indicating that attenuated DHCR7 levels and 7-DHC accumulation allow higher flux into the vitamin D3 pathway [43]. Such data points out that DHCR7 might act as a biological switch between vitamin D3 and cholesterol synthesis [40].
Where does vitamin D come from?
Vitamin D was initially used by early single-celled organisms [44]. It remains unknown why these primitive creatures produced ergosterol and vitamin D2, but three possible hypotheses have been suggested: Ergosterol can effectively absorb UVB. Thus, it can protect UV-sensitive macromolecules such as DNA, RNA, and protein. Vitamin D2, which is synthesized after UVB is absorbed, also absorbs light of 260 nm wavelength, which is the light absorbed by DNA and RNA. In this way, vitamin D2 also protects DNA and RNA against UVB [37, 45]. Finally, Tachysterol2, which is formed as a result of the encounter of vitamin D2 with UVB, absorbs rays of 282 nm wavelength, which coincides with the UV rays absorbed by proteins [37, 45–47]. According to this idea, organisms that used sunlight for photosynthesis in the early stages of evolution needed to protect their UVB-sensitive molecules from the harmful effects of UVB radiation [37]. Ergosterol, vitamin D2, and their photoproducts have been ideal UVB protectors and sunscreen. On the other hand, the amount of vitamin D2 and photoproducts acts as a photochemical signal (actinometer) and states how much UVB the organism was exposed to. The organism, which can monitor the amount of UVB it receives in this way, also has a warning mechanism to return to the depths of the sea and protect it from the sun after receiving sufficient UVB[37, 45].
In addition, according to another proposed mechanism, if ergosterol is located in the lipid bilayer of the plasma membrane, the formation of vitamin D2, which is more flexible with UVB and possibly released into the extracellular space, may change the membrane permeability and lead to possible pore formation, allowing ions such as calcium to enter and exit the cell. This mechanism may explain why vertebrates, including humans, need sunlight to maintain calcium metabolism [37, 45–49]. Along with bone calcification and formation, this molecule also participates in many cellular events such as the immune system, cellular growth, and differentiation [44].
The most critical step in the formation of active vitamin D is the conversion of 7-dehydrocholesterol to vitamin D by UVB. The central importance of this step can be highlighted by the gradual depigmentation that modern humans underwent during their journey out of Africa [44] approximately 194-175,000 years ago [50].
THE EFFECT OF VITAMIN D AND ITS DEFICIENCY IN CNS WITH REGARD TO BOTH IN VITRO AND IN VIVO STUDIES
Effect of vitamin D on the central nervous system (CNS)
Recent studies have shown that vitamin D has numerous effects on the nervous system. These effects include the regulation of key survival mechanisms such as neurotrophic factor production, regulation of oxidative stress, calcium homeostasis, and immune system functions [2, 51–56]. The relationship between vitamin D deficiency in the early stages of life and diseases such as autism spectrum disorders and schizophrenia, has been tried to be explained by the vitamin D deficiency on neuronal differentiation, axon connections, dopamine ontogeny, brain structure, and function [57]. On the other hand, studies are showing that vitamin D deficiency triggers premature aging [58], causes enlargement of the lateral ventricles, decreases nerve growth factor, decreases the expression of genes related to neuronal structure, deteriorates brain development, and therefore causes changes in the brain of the adult individual [14]. Vitamin D has been suggested as protector against aging [59]. It has also been suggested that vitamin D deficiency during development leads to changes in behavioral patterns against agents that stimulate or block dopamine release in adults [60, 61]. Although few human studies also suggest that low blood 25(OH)D levels are associated with mood disorders, dementia, mild cognitive impairment, Parkinson’s disease (PD), AD, and cognitive decline [62–70].
Benefits of vitamin D supplementation in cell culture and animal models
AD is a progressive neurodegenerative disease that begins long before its symptoms arise [71]. It is the most common cause of elderly dementia [11]. AD-type pathology includes extracellular amyloid plaques and intracellular protein aggregation neurofibrillary tangles [72]. These protein aggregations cause adverse events like disruption in signal transduction, axonal transport, mitochondrial function and neurotrophic factor metabolism, induction of oxidative stress, and alteration of neuronal ion homeostasis, leading to neurodegeneration [72–75]. Our group’s previous research, AD-related cell culture experiments, and other animal studies revealed the apparent benefits of vitamin D administration like regulating oxidative stress immune response related mechanisms, ion channels, AD pathology related enzymes, amyloid-β (Aβ) production, preventing Aβ-related toxicity, cell membrane damage, and increasing neuronal survival [30, 77]. Relatively known aspect of vitamin D is that it is a significant contributor in regulating many cytokines [78, 79] in different cell types in the immune response and therefore was also linked with multiple sclerosis. How vitamin D contributes to neuro-immunomodulation has been meticulously reviewed by other researchers [80, 81]. Vitamin D-supplemented AD mouse models showed improved inflammatory and immune gene expression profiles [82]. Vitamin D stimulated cytokines and macrophages and increased Aβ clearance in AD cases [83, 84] and in aged rats [85]. On the other hand, our in vitro studies conducted on primary hippocampal or cortical neuron cultures indicated that 1,25(OH)2D3 treatment attenuated CaV1.2 [56] and CaV1.3 [55] iNOS [54, 86] expressions, increased NGF [87], resisted Aβ toxicity [7, 87], regulated most of the members of amyloid processing mechanism including presenilin 1, presenilin 2, NICASTRIN, ADAM10, BACE1, and amyloid-β protein precursor (AβPP), attenuated Aβ production [7], and resulted in enhanced neuronal survival and neuroprotection [7, 87] (Fig. 1). In addition, it has been reported that 1,25(OH)2D3 administration stimulates Aβ phagocytosis and clearance, and its excretion from the brain to the blood [83, 88–90]. Furthermore, it has been shown that high vitamin D and fish oil consumption can prevent neurodegeneration [89]. It has been shown that 1,25(OH)2D3 increases neurite outgrowth in embryonic hippocampal neurons [52, 91] and can change cholinergic, dopaminergic, and noradrenergic neurotransmitter systems in the CNS [57]. It has effects on choline acetyl transferase enzyme [92], cholinergic receptors [93], dopamine receptors [94, 95], and tyrosine hydroxylase enzyme [96, 97]. It has been shown that vitamin D binding protein, or group-specific component globulin (GC-globulin), which is the main carrier of vitamin D in the blood, also prevents Aβ accumulation and Aβ-induced cell death [98]. The transcriptomic data of mixed neuron-glial cell culture showed that neurodegeneration, neuroinflammation, or brain development-related proteins such as hydrogen sulfide-producing enzyme cystathionine beta-synthase, triglyceride hydrolysis enzyme lipoprotein lipase, a membrane-bound synaptic protein carbonic anhydrase 14, a neuronal and epithelial glutamate transporter solute carrier family 1, a free cholesterol into cholesteryl ester converting enzyme lecithin-cholesterol acyltransferase, an extracellular matrix protein that interacts with the amyloid precursor Fibulin 1 were effected by 1,25(OH)2D3 administration [99]. In another study, 117 genes known to be involved in AD pathology have been examined in a mouse model having a 20% –30% vitamin D hypovitaminosis. The mRNA expression of Plat, Gnb5, Nep, Park7, Psmb5, Casp4, Snca, and Acat1 genes which are related to AβPP metabolism, oxidative stress, inflammation, lipid metabolism, G proteins, and neurogenesis were found to be significantly changed in vitamin D deficient mouse brains [100].
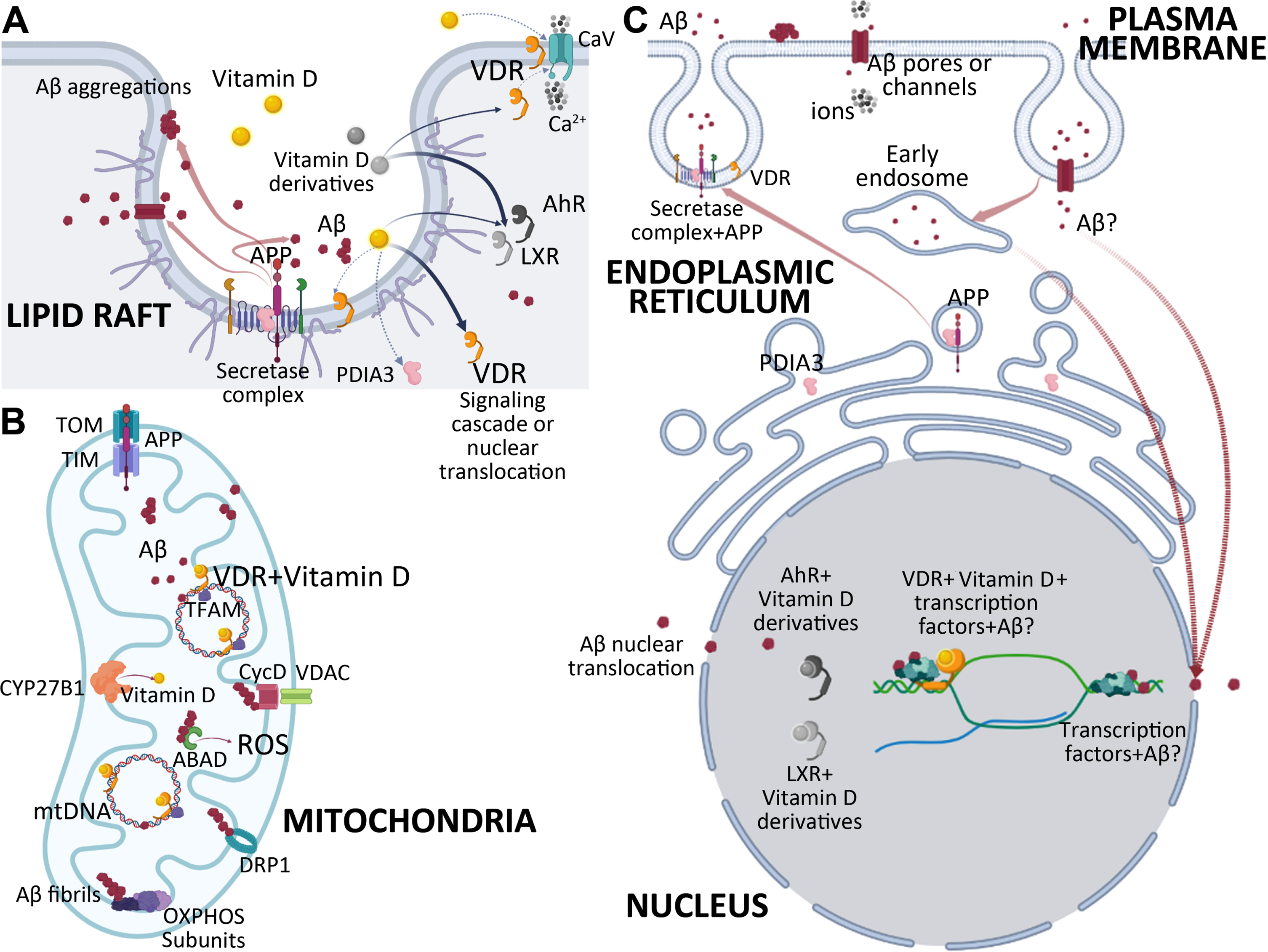
The summary of vitamin D, vitamin D receptors, and Aβ action in CNS: A) Lipid rafts: Lipid rafts suggested including both VDR receptors and AβPP processing enzymes. α-, β-, and γ-secretases are located on lipid rafts where they sequester AβPP to produce Aβ or other fragments. Our studies indicated that this complex might include VDR too. Aβ may accumulate on lipid membranes without an enzymatic reaction forming protein aggregations, Aβ pores, or channels, disrupting the membrane integrity and selective permeability. VDR and Aβ both have effects on CaVs, thus on intracellular Ca2 + levels. Vitamin D mainly acts through VDR/RXR heterodimers, whereas vitamin D derivatives prefer AhR or LXR. The action of the vitamin D may involve activating signaling cascades like PLC, PKC, MAPK/ERK, AKT/mTORC, Wnt, β-catenin, or transcriptional regulation via VDR. B) Mitochondria: Mitochondria is the main site that involves cytochrome P450 enzyme CYP27B1 which is involved in vitamin D synthesis. AβPP is reported to be translocated into mitochondria via TOM-TIM translocators. Aggregation of Aβ peptide is reported on mitochondrial membranes. This accumulation is suggested to interfere with OXPHOS activity. Studies also indicated that Aβ affects the expression levels of OXPHOS subunits. Aβ interacts with CycD-VDAC, DRP1, and ABAD, which in turn disrupts mitochondrial activity and energy metabolism. Our studies indicated that the vitamin D receptor regulates the transcription of mitochondrial DNA and directly interacts with mitochondrial DNA and TFAM. This regulation may also include or be interfered with Aβ. C) Plasma membrane-ER-Nucleus: The plasma membrane is one of the sites where AβPP and AβPP processing enzymes are located. Recent studies reported that AβPP processing requires the invagination of AβPP-containing plasma membranes and cleavage of AβPP to produce Aβ in the endocytic vesicles. These vesicles are suggested to release their ingredients by exocytosis in response to stimulation. The Aβ monomers and oligomers are released to extracellular space. Aβ aggregates on lipid membranes may form pores or channels disrupting the membrane integrity and selective permeability. Released Aβ may be internalized by endocytosis. These endocytic vesicles may lead to endosomes. Endoplasmic Reticulum-Golgi-Plasma membrane vesicular traffic includes subcellular structures where AβPP processing occurs. PDIA3 was reported to be interacting with AβPP directly in this axis. Translocation of Aβ to the nucleus via nuclear pores is also reported. Studies indicated that Aβ binds to DNA sequences to regulate gene expression. VDR is a well-known transcription factor. Vitamin D/VDR transcriptional regulation may also involve Aβ.
Vitamin D deficiency and vitamin D supplementation in human studies
Vitamin D Standardization Program supplied valuable data about the prevalence of vitamin D deficiency and its detection protocols and suggested the deficient serum level of 25(OH)D to be <30 nmol/L [101]. Vitamin D deficiency reported to be concerning European, North American, and dark-skinned ethnic groups [102–105]. Additionally, vitamin D deficiency reported to be seen significantly more frequent in the elderly then the children [104, 105]. Low levels of serum 25(OH)D in AD, PD, mood disorders, and cognitive decline and motoric cognitive risk syndrome cases have been reported [5, 106–111]. Some longitudinal studies have confirmed the association between low 25(OH)D levels and cognitive decline in the elderly [68]. A recent one supported the hypothesis by showing the increased risk of developing AD in vitamin D deficient population [108]. A positive correlation between serum 25(OH)D levels and Mini-Mental State Examination (MMSE) scores has also been reported [69]. Annweiler et al. suggested the involvement of vitamin D deficiency in the prodromal stages of dementia [64] and its potential to be a predictive factor for executive dysfunction, especially with regard to mental shifting, information updating, and processing speed [112]. A meta-analysis noted lower serum 25(OH)D levels in AD cases compared to healthy subjects [113]. Another one indicated significant associations between vitamin D deficiency and both dementia and AD. The level of association positively correlated with level of vitamin D deficiency [114]. Yet one analysis failed to show the association [115]. One meta-analysis indicated that AD decreased continuously along with the increase of serum 25(OH)D [116]. A longitudinal study that followed the cases for 30 years indicated an association of low plasma 25(OH)D levels with increased risk of AD [117]. Our study in 2016 indicated that vitamin D deficiency might pose a greater risk for APOE ɛ4 non-carrier AD cases [5], and our results were confirmed in a larger cohort in Canada [118]. Recently, Kiderman et al. indicated that patients with insufficient and deficient vitamin D levels were diagnosed with dementia at younger ages [119].
Recently, variations in serum 25(OH)D concentration thresholds for optimal health meticulously reviewed by Grant et al. and indicated that concentrations to support health and wellbeing should be above 30 ng/mL, in some certain disease conditions above 40 or 50 ng/ml [120]. Though, we should say that researchers and physicians are still not sure about the optimal serum 25(OH)D concentrations for general health and even not for bone health [121]. This is due to its variations in race, ethnicity, and stage of life and perhaps some chronic disease conditions [122–124]. Other factors like geography, seasons, cultural features, and nutrition are also suggested to be the case for vitamin D deficiency. Besides, the efficiency of vitamin D synthesis in the body is known to be dependent on the pigmentation of the skin, geographic latitude, the sunlight, and the individual’s age [125]. Aging itself affects vitamin D production [126]. Vitamin D deficiency is highly frequent in obese subjects [127] and obesity is also a risk factor for AD [128]. Obesity, adipose tissue, lipid metabolism, leptin, adipokines, and their relation to AD [129–134] and vitamin D [135–137] should not be ignored. Given that there is a debate on what would the optimal level be on every platform [138–141]. Besides, vitamin D supplementation is not able to increase serum 25(OH)D concentrations linearly [122], although we managed to define a formula to predict the increase of serum 25(OH)D levels with a 14-day long regiment, when we were working with COVID cases. Our treatment protocols in our COVID study indicated that the elevation in a patient’s serum 25(OH)D levels within 14 days could be predicted with the formula “y = 8.63 ln(x) + 13.66”, where x = the initial serum 25(OH)D level and y = the predicted serum 25(OH)D levels in 14 days after treatment with that regiment [142]. The study included a 2-week treatment. The week starts with a 100,000 IU oral dose of cholecalciferol followed by 10,000 IU daily supplement for 6 days for cases with serum 25(OH)D level <12 ng/mL (total dose of 320,000 IU), 5,000 IU daily supplement for 6 days for cases with serum 25(OH)D level 12–20 ng/mL (total dose of 260,000 IU), 2,000 IU daily supplement for 6 days for cases with serum 25(OH)D level 20–30 ng/mL (total dose of 224,000 IU) [142]. The study indicated a 2.14 times reduction in mortality rates of COVID-19 cases with comorbidities after 14 days of vitamin D supplementation, which aimed to increase serum 25(OH)D levels above 30 ng/ml and reached that goal within 14 days [142]. The comparison of the recommended daily intakes by the different national health authorities was reviewed by Pilz et al. [143].
Vitamin D and its analogues regarding to the differences in molecular mechanisms and their use in AD treatment were recently reviewed [144]. Clinical trials and vitamin D intervention studies in neurological disorders were meticulously reviewed by Plantone et al. [26]. The team also reviewed vitamin D treatments in AD [145]. Although some studies indicate a lack of benefit [146–150], others noted promising results [151–153]. Annweiler et al. reported an average of four points increase in the MMSE scores of AD patients with a vitamin D and memantine combined treatment protocol [153]. However, regardless of their results, we should note that each trial varies by means of the cases included or the treatment protocols utilized. Thus, vitamin D intervention in AD requires further investigation. As we mentioned before, vitamin D is a secosteroid hormone that should be kept at adequate levels in every stage of human life to establish a high cognitive reserve [8, 154]. Nevertheless, given those years of untreated vitamin D deficiency during a person’s life, someone should not expect to treat the results of this deficiency with one or two years of intervention. So, some significant issues should be kept in mind. First, such intervention studies should include a clinically well-defined and homogenous set of patients; rather, cases at the early stages of the disease, perhaps with clinical dementia rating (CDR) 0 or 1, if ATN scale [155] provided then A–T–(N)–, A+T–(N)–, or A+T+(N)–might be preferred. The goal should be to investigate long-term benefits, possibly within one or two years. Besides recording the endpoint effect like MMSE score or other cognitive functions, alterations in cerebrospinal fluid (CSF) or plasma biomarkers or neuroimaging data should be included. At this point we should note that Soares et al. reported that participants with higher CSF levels of 25(OH)D exhibited reduced CSF tau and phosphorylated tau levels [156]. The baseline serum 25(OH)D levels should be recorded, and the treatment regimens should be adjusted according to the baseline vitamin D levels [142].
THE MECHANISM OF VITAMIN D/VITAMIN D RECEPTORS AXIS
Vitamin D receptor
Although the efficacy of enzymes that activate vitamin D, such as 1α-hydroxylase (CYP27B1) in tissues other than the kidney, is controversial [157, 158], VDR and 1α-hydroxylase were found to be abundantly expressed in many regions of the CNS. Therefore, it is particularly important that they are expressed widely, especially in the areas affected by neurodegenerative diseases [2, 159].
VDR is one of the 1900 transcription factors encoded in the human genome and is expressed in many human tissues and different cell types [44] (Fig. 1). The VDR carries a highly conserved DNA binding site and a constitutively conserved ligand binding site, which are the main structural characteristics of nuclear receptors. The DNA binding site associates with the particular consensus sequences in the major groove of DNA. VDR uses a partner protein to bind more effectively to its target genes. This protein is another nuclear receptor, the retinoid X receptor (RXR) [44]. When VDR behavior is modeled, it has been found that VDR acts as one of the transcription factors in the concept of cyclic gene regulation, which fluctuates between the on and off states. The effect of cholecalciferol on VDR expression demonstrated by the study utilize it as the supplementation of 90 healthy adult monozygotic twins for 2 months and the treatment resulted in sixty times increased VDR gene expression [160]. The genomic function of the VDR is controlled by epigenetic changes in receptor binding sites [161]. The VDR keeps its level under control by proteosome-mediated receptor reduction. Although VDR activity is better characterized for positive gene regulation, it is also involved in VDR gene suppression and induction. In the absence of the ligand, the VDR coexists with corepressors in the nucleus at the basal level and suppresses transcription [161, 162].
VDR works with transcriptional coactivators such as, CBP/p300 (works as transcriptional co-integrator with histone acetylase) [163, 164], SNW1/NCOA62 [165], CCND3 [166, 167] and corepressors such as HR [168].
Besides these traditional transcriptional regulatory roles of the VDR, it also plays a very broad role in mRNA control. Proteins that interact with the VDR, such as SRPK1 or SNW1, are involved in RNA splicing. Besides, VDR is associated with many signaling pathway regulators: Wnt signaling pathway regulators (NKD2), PKC substrates (PRKCSH), p53, SMAD3. It also binds with many corepressors such as NCOR1, NCOR2/SMRT, TRIP15/ALIEN, and DREAM [161].
Transcriptomic studies using microarray showed that VDR regulated transcripts were high in number, of different types, and highly heterogeneous by cell type and time [161]. On the other hand, with the discovery of miRNAs, it has been shown that VDR can also control many miRNAs. ChIP-Seq studies indicated that the VDR binding sites show a great diversity according to the cell types. Likewise, many VDR response elements that are not known to be used under normal conditions in nature are located in genes and the underlying mechanism is unknown [161].
According to the latest data of Genecode 2022 (February 2023, GRCh38.p13) Version 43, the human genome contains 19.393 protein-coding genes. RNA processing and editing results in 252.913 gene transcripts (https://www.gencodegenes.org/human/stats_43.html). For VDR, published results from VDRChIP-seq datasets and ENCODE are quite surprising [44]. Currently, VDRChIP-seq information is available for the GM10855 and GM10861 lymphoblastoid cell lines [169], LPS polarized THP-1 macrophage-like cells [170], LS180 colorectal cancer cells [171], and LX2 hepatic stellate cells [172]. In these six VDRChIP-seq data sets, 21,776 non-overlapping VDR binding sites with 250 bp intervals were detected [44]. 67% of these VDR binding sites are specific to the cell type. Only 54 of them are common to the six VDRChIP-seq datasets. Repeated analyzes with these datasets show that there may be 1000–10,000 genomic VDR binding sites per cell type [44]. Again, the data obtained from these studies showed that VDR combined with different coactivators and corepressors in the presence and absence of ligands [44].
Protein disulfide isomerase A3 (PDIA3)
It has been suggested that the other suggested receptor of vitamin D initiates the cellular response to vitamin D by signaling mechanisms or transcriptional regulation [173] and can be found together with the VDR in the plasma membrane [174]. It is an endoplasmic reticulum protein, protein disulfide isomerase A3 (PDIA3) or as it has been named, P58, ER60, ERp57, ERp60, ERp61, GRP57, GRP58, and vitamin D membrane-associated, rapid response, steroid binding protein (1,25-MARRS) and was suggested to be a membrane receptor for vitamin D (Fig. 1). PDIA3 is belong to thioredoxin (TRX) superfamily which is a large family of oxidoreductase enzymes that are common and abundant in the cell [175]. PDIs are mainly localized in the lumen of the endoplasmic reticulum (ER), and they catalyze thiol-disulfide conversions, formation (reduction), oxidation and isomerization of disulfide bonds, which are critical during the folding of newly synthesized proteins passing through the secretory pathway. They can act as important molecular chaperones that play physiological or pathophysiological roles and help prevent the aggregation of unfolded or misfolded proteins as part of a quality control system [176, 177]. Despite the presence of an ER retention sequence at the C-terminus of the protein sequences of PDI family members, some PDIs are beginning to be shown to be located in other subcellular compartments, including the cytosol, mitochondria, nucleus, plasma membrane, and extracellular space [176]. Although PDIA3 has been suggested as a membrane receptor for vitamin D, our results with live-cell labeling have shown that its level in cortical neuron membrane is very low compared to VDR and it is abundantly localized intracellular [178].
Since PDIA3 is still very new in the field of vitamin D or neurodegeneration, only a few studies have examined the mechanisms of PDIA3-vitamin D in the brain, and the presence and functions of PDIA3 in cortical neurons have been reported [54, 179].
The full-length AβPP was reported to interact with PDIA3 in the ER [180]. PDIA3 was reported to be altered significantly in hippocampus of Aβ1–40-treated rats [181] and in the brains of 3×Tg-AD mice [182, 183]. The first report that showed the increased amyloid production in PDIA3 silenced cortical neurons was done by our group. In that study we suggested that PDIA3 absence might result in a loss of control over AβPP processing in the ER or the early secretory pathway [7] (Fig. 1). In another study we showed that CaV1.3 and CaV1.2 were attenuated with vitamin D. Interestingly only CaV1.2 mRNA expression responded to VDR absence; however, CaV1.3 responded to PDIA3 absence [54].
Furthermore, newly identified pathogenic PDIA3C57Y expression was reported to cause axonal disorganization and skeletal abnormalities in zebrafish embryos; impaired synaptic plasticity and memory consolidation in the mouse hippocampus. The study showed that this mutant form of PDIA3 has decreased catalytic activity and forms disulfide-cross-linked aggregates and therefore it drives defects in cell adhesion and actin cytoskeleton dynamics and reduces neuritogenesis [184].
DHCR24 have neuroprotective effects including resistance to oxidative stress by reinforcing the membranes with synthesizing cholesterol and by inhibiting caspase 3 and is suggested to associate with AD [185, 186]. Cellular cholesterol levels have an effect on vitamin D production, and high cholesterol concentrations can convert 7-DHC to vitamin D synthesis instead of going through the final stage of cholesterol synthesis [187]. Recently we showed that vitamin D administration decreased DHCR24 mRNA and protein levels. When we silenced the receptors to understand whether this effect was due to VDR or PDIA3, we found that the pathway was probably mediated by PDIA3. This study showed that both vitamin D and PDIA3 are involved in cholesterol metabolism [40]. It is particularly important because cholesterol is necessary for steroid synthesis, neural transmission, axonal development, and synapse formation [188, 189].
In addition, it has been reported that PDIA3 and calreticulin inhibit Aβ aggregation in the CSF [179], stimulate axonal growth and axon regrowth in toxic conditions [78]. Although PDIA3 seems to stand in a very important place, we need more studies to understand how much of the functions of PDIA3 are directly related to vitamin D.
Vitamin D derivatives and their receptors
The other metabolites of vitamin D and the other receptors with which these metabolites interact also complicate the event— for example, a transcription factor Aryl hydrocarbon receptor (AhR). Although AhR is known for its ability to mediate the effects of environmental toxins such as dioxin, critical physiological roles in the developing nervous system of invertebrates and vertebrates of this evolutionarily conserved molecule were reported recently [190]. AhR was also associated with neuronal subtype specification, cell and axon migration, axonal branching, dendritic morphology, and cerebellar granule cell neurogenesis in different organisms [191–194]. Developmental roles seem to be different from the adult aging process. There are controversial results in different organisms, including humans, and assessed AhR as both promoting and preventing factor for aging [195]. Studies in human keratinocytes showed it is associated with various cutaneous immunologic processes, chemical carcinogenesis, development of non-melanoma skin cancer, photocarcinogenesis, and skin inflammation [190, 196], which are almost opposite to the effects of VDR. The UVB-induced stress response involves AhR activation and the hormonally active form of vitamin D, and non-classical vitamin D derivatives are produced by UVB. It is found that especially vitamin D derivatives can bind to AhR (Fig. 1). The cross-talk between VDR and AhR is a new field, and one study in keratinocytes showed differential but orchestrated regulatory properties of these transcription factors [196]. Recently liver X receptors (LXRs) were identified as a new receptor for several D3-hydroxyderivatives, including 1,25(OH)2D3 in murine dermal fibroblasts [197]. LXR is a nuclear receptor that works like VDR and heterodimerizes with RXR. Single-cell transcriptomic data showed that the neurogenesis and differentiation of human embryonic stem cells derived from midbrain dopaminergic neurons were enhanced by activation of LXR [198]. LXRβ in astrocytes of the medial prefrontal cortex was suggested to be critical in the regulation of synaptic transmission [199]. LXR knock-out models also suffer from severe brain abnormalities [200]. The cross-talk between VDR and LXR is not studied as far as we know (Fig. 1).
Vitamin D receptors and neurodegeneration
The initial study that pointed out the involvement of VDR in AD was provided by Sutherland et al. in 1992 [201]. They reported attenuated levels of VDR mRNA in the hippocampal neurons of AD cases [201]. Following this study, several genetic studies reported a novel AD risk locus on chromosome 12 [202–205]. We thought that these studies were critical since the risk locus include the VDR gene. Our initial hypothesis was relatively simple: if vitamin D is known for maintaining calcium homeostasis in the human body, it should also function in brain, a major organ that utilizes calcium. Furthermore, the study provided the first evidence of a genetic relation between vitamin D/VDR and AD [206], thus in brain function. Studies confirmed this relationship at the genetic level [5, 207–212] and biochemical level [62, 106]. Some Mendelian randomization analyses also confirmed the associations of serum 25(OH)D concentrations with AD [213, 214]. Vitamin D regulates neurotrophic factors and voltage-sensitive calcium channels in the brain [3, 215]. The initial molecular studies of our group targeted NGF, CaV1.2, and CaV1.3. We found that Aβ induces neurodegeneration by inducing the expression of CaVs and altering NGF synthesis [56]. Besides, we showed that Aβ significantly suppressed VDR expression [56]. This significant result also gave a chance for a possible molecular explanation for the observations of Sutherland study. The following findings of our group indicated that Aβ induces 24OHase [76], the enzyme that accelerates vitamin D catabolism and the attenuates of 1α-hydroxylase [75], is responsible for the production of the active form of vitamin D. We speculated that cognitive functions may require high amount of vitamin D, since we detected high expression of VDR and the 24OHase enzyme in hippocampal neurons [30]. Our results also revealed that there is a cross-talk between Aβ pathology and vitamin D action. Similar to our study that showed VDR suppression in Aβ treated neurons a recent study reported decreased VDR level in 2xTg-AD mice. In same study, peroxisome proliferator activated receptor (PPARγ) coactivator-α (PGC-1α) was also reduced in this transgenic mice model and it was suggested PGC-1α could serve as a coactivator for VDR and prevent oxidative damage [216]. So the question was whether the vitamin D-VDR/PDIA3 axis could be involved in a novel model of neurodegeneration. We disrupted the vitamin D pathways either via vitamin D-VDR or vitamin D-PDIA3 by siRNAs, and models resulted in the induction of CaV1.2 and CaV1.3, iNOS, and attenuated NGF. All these alterations previously reported be potentially stimulate Aβ accumulation, thus neurodegeneration [30, 55]. The data indicated that either Aβ–or VDR absence induced similar amounts of iNOS and CaV1.2 mRNAs in neurons [54–56, 87]. Given that these data seemed secondary to the mechanism of AD-type neurodegeneration, the following question was more direct. Does the vitamin D-VDR/PDIA3 pathway affect the enzymes which regulate Aβ production? The targets of our study were AβPP processing cascade in AD pathogenesis [217]. We showed that vitamin D and/or its receptors modulate the expression of secretases (presenilin 1, presenilin 2, NICASTRIN, ADAM10, BACE1) and the AβPP in a time-, concentration-, and receptor-dependent manner [7]. Furthermore, 1,25(OH)2D3 administration decreased intracellular Aβ1–42 production and secretion [7]. Similarly, Aβ production decreased, whereas Aβ degradation increased in response to vitamin D2 and D3 analogs in neuroblastoma cells or brains of vitamin D-deficient mouse [218]. Today, accumulating studies are supporting our findings and pointing out a regulatory role for vitamin D-VDR and PDIA3 in the brain development and neurodegeneration [82–84, 219–222].
On the other hand, a recent study in human ApoE-expressing rat astrocytes showed that ApoE2 activates VDR in Aβ42-treated astrocytes and 1,25(OH)2D3 treatment rescues inflammatory gene expression in human ApoE4-expressing astrocytes. The authors proposed that ApoE4 promotes inflammation while ApoE2 protects against neuroinflammation in AD, acting via the VDR signaling [223]. Another study reported that the folate transport systems are regulated by VDR. Mutations in folate transport system genes cause early childhood neurodegeneration. In different in vitro blood-brain barrier model systems, VDR was shown in the direct regulation of this folate transporters [224]. In addition, VDR and a VDR target, P-glycoprotein (P-gp), encoded by MDR1a were reported to attenuate in 6-OHDA and α-synucleinopathy PD mouse models. In this study, α-synuclein preformed fibrils induced P-gp downregulation in HUVECs and the study showed these down-regulation depends on vitamin D, and vitamin D treatment rescue the adverse effects of fibrils [225].
Other functions of VDR in neurodegeneration
VDR and mitochondria
Neurons are energy-demanded cells and producing their energy from the mitochondrial oxidative phosphorylation (OXPHOS) system. Disruption of mitochondrial function is related to be neurodegenerative disorders [226, 227]. Aβ fragments was reported to be transported to mitochondria in pathological conditions [228]. Aβ1–42 plaque density was elevated in a transgenic AD mouse model which has disrupted proofreading function of mitochondrial DNA polymerase γ [229]. The expression of nuclear DNA encoded OXPHOS subunits was reported to decrease in the early stages of AD [230]. Additionally, the expression of mitochondrial DNA (mtDNA) encoded OXPHOS genes was changed in mild cognitive impairment and AD patients compared to healthy subjects [231]. Recently, we reported that the expression of some mtDNA-encoded genes and mitochondrial quality genes such as PARKIN and PINK1 are changed in PD cases compared to the healthy subjects [232]. Our group recently reviewed the DNA binding capacity of Aβ, speculated that Aβ or α-synuclein can be a cofounder in mtDNA gene expression and might have an ability to bind to mtDNA [13] (Fig. 1).
According to transcriptome and proteome results, vitamin D deficiency was suggested to have an effect on brain energy metabolism [233] and to be associated with increased muscle atrophy by reactive oxygen species generation and disruption of mitochondrial function [234]. In human skeletal muscle cells, 1,25(OH)2D3 treatment effects the expression of 83 nuclear mRNAs encoding mitochondrial proteins and elevates mitochondrial oxygen consumption rate [235]. Similar findings reported in primary rat granulosa cells and 1,25(OH)2D3 was suggested to increase the expression of mitochondrial biogenesis-related proteins, ATP production, and cellular oxygen consumption [236]. Developmental vitamin D deficiency was reported to be related with the production of six nuclear DNA encoded mitochondrial proteins in rat nucleus accumbens [237].
Furthermore, Silvagno et al. showed the presence of VDR in the mitochondria of human platelets, differentiated megakaryocytes and human keratinocyte cell line for the first time [238, 239]. VDR silencing was reported to cause impairment of mitochondrial integrity and cell death [240].
VDR is a transcription factor (TF) (Fig. 1). In our recent study, we demonstrated that VDR might serve as a TF for mtDNA and we showed the co-localization of VDR with mitochondria and the binding of VDR to mtDNA D-loop site in several locations. Furthermore, our findings indicate the possible interaction between VDR and TFAM (Fig. 1). Our data shows for the first time that VDR is able to regulate mtDNA transcription and interacts with TFAM in different neuronal cells and human brain [241]. We suggested that an imbalance of this secosteroid hormone or its receptor has crucial roles in the development and progress of mitochondrial dysfunction-related diseases like neurodegenerativedisorders.
VDR on plasma membrane
The complex effects VDR silencing or 1,25(OH)2D3 applications at different doses and times in our studies and the effect of Aβ on VDR suppression, which we found in our previous studies, led to the idea that all of the effects we observed cannot be explained by transcriptional regulation alone. This proposition prompted us to investigate the possibility of VDR being present in the neuron plasma membrane as a possible regulator of signal-propagating protein or secretase complex (Fig. 1). Limited number of studies have suggested that the classical VDR, which plays a role in gene transcription, may also be associated with or located near the caveolae in the plasma membrane of osteoblasts [242, 243]. Bartoccini et al. demonstrated that the VDR is localized in lipid-rich microregions in nuclear membranes, which are structurally equivalent to microregions found in the plasma membrane of developing hippocampal neurons [244]. Rapid membrane response is a cellular response that provides rapid calcium flow through the membrane, and calcium flow in neurons plays a critical role in neuron firing and memory formation. One of the primary roles of vitamin D is to regulate calcium and phosphate metabolism. Considering these reasons, we hypothesized that the VDR could also be found in the neuron plasma membrane and function as a part/regulator of various protein complexes. The possibility of the VDR, classified as a nuclear hormone receptor, to undertake such a role at the plasma membrane is an area that has yet to be investigated. To test this hypothesis, we marked primary cortical neurons with the anti-VDR antibody by live-cell surface staining technique [245], and determined that the VDR was in the neuronal plasma membrane [178]. Using this technique, our team demonstrated for the first time that VDR can be found on the surface of primary cortical neurons (Fig. 1). We also detected the presence of VDR on the plasma membrane in SH-SY5Y cells and observed that the localization of the VDR on the plasma membrane is not limited to primary cortical neurons. The same study also showed that the possible co-localization VDR with proteins involved in amyloidogenic pathway[178].
Cross-talk between Aβ and VDR
Previously we mentioned that our results showed suppression of VDR by Aβ. Recently we hypothesized and reviewed the possible mtDNA binding capacity of Aβ and possible interaction between Aβ and TFs [13]. Although we are still working on this hypothesis, the emergence of this hypothesis came from some data we obtained while investigating the relationship between VDR and Aβ. Initially, it has been reported that Aβ translocation to the nucleus and binds to DNA from its N-terminal region, altering the expression of certain genes [246–250]. In several studies conducted by our group, its effect on the expression of genes related to vitamin D metabolism (VDR, 24OHase, 1α-hydroxylase) as well as genes related to neurodegeneration (APP, Presenilin 1, Presenilin 2, BACE-1, tau, GSK3β, CDK5, Trem2, APOE, iNOS, CaV1.2, and NMDRAs-Grin1, Grin2a, Grin2b, Grin2c, Grin2d, Grin3a) has been shown [30, 76]. In addition, in primary cortical neurons, we have shown that Aβ can migrate from the cytoplasm to the nucleus in response to the certain antibiotic doses [7]. As a result of the analyzes that we made with the FPClass PPI program, AβPP or its fragments might interact with TFs and when we combined the data that we gathered from our expression studies, FpClass data and TRRUST database, we determined possible TFs that might be interacting with Aβ as follows: ETS2, JUN, JUND, SP1, STAT1, STAT3, TBP, SMAD3, SNAl1, NFKB1, RELA, ELK1, ATF4, TFCP2, APC, FOS, FOSL2 [13]. Supporting single-soma transcriptomic data showed that JUN and ATF4 effected in tangle-bearing neurons in AD [251]. Interestingly in TRRUST database 19 different TFs (https://www.grnpedia.org/trrust/result.php?gene=vdr&species=human&confirm=0) including JUN, SNAl1, and SMAD3 were proposed for transcriptional regulation for VDR [252–255]. Whether it interacts directly with DNA or interacts with TFs, this information indicates that Aβ has an effect on gene expression and also might have a role in VDR gene expression. Since the data of Sutherland et al., confirmed by a few studies including by us and other groups, showed that VDR expression is reduced by Aβ pathology, it reinforces the idea that there is a cross-talk between VDR-Aβ (Fig. 2).
On the other hand, miRNAs might be another issue for Aβ-VDR cross-talk. Some studies suggested that VDR mRNA expression is regulated by mir-27b-3p [256], let-7a-5p [257], or mir-125b-5p [258]. CYP2R1 encoding 25OHase, which hydroxylates of vitamin D [1], can be suppressed by let-7a-5p [259]. CYP27B1 suppression by vitamin D was reported in intestinal cells [260]. CYP27B1 expression suggested to be regulated by mir-26b-5p and mir-31b-5p [259, 261]. Our studies indicated a possible involvement of let7a-5p in Aβ-VDR crosstalk [262]. Aβ1 - 42 induced elevation of let-7a-5p might underline the possibility of Aβ to disrupt vitamin D synthesis at the earliest steps. Besides, our study indicates that possible involvement of mir-26b-5p and miRNA31b-5p in CYP27B1 regulation [262]. Studies have suggested the regulation of 24OHase by mir-125b-5p [263], mir-26b-5p [261], mir-215-5p [264], and mir-192-5p [264]. Our study on the other hand, indicated that elevation in vitamin D levels can induce CYP24A1 expression and downregulate mir-26b-5p and mir-192-5p. That study also showed that Aβ downregulated the expression of mir-125b-5p and mir-26b-5p [262]. At this point we have to note that Aβ1 - 42 has the ability to suppress the mRNA expression of 1αOHase in neurons [76] (Fig. 2).
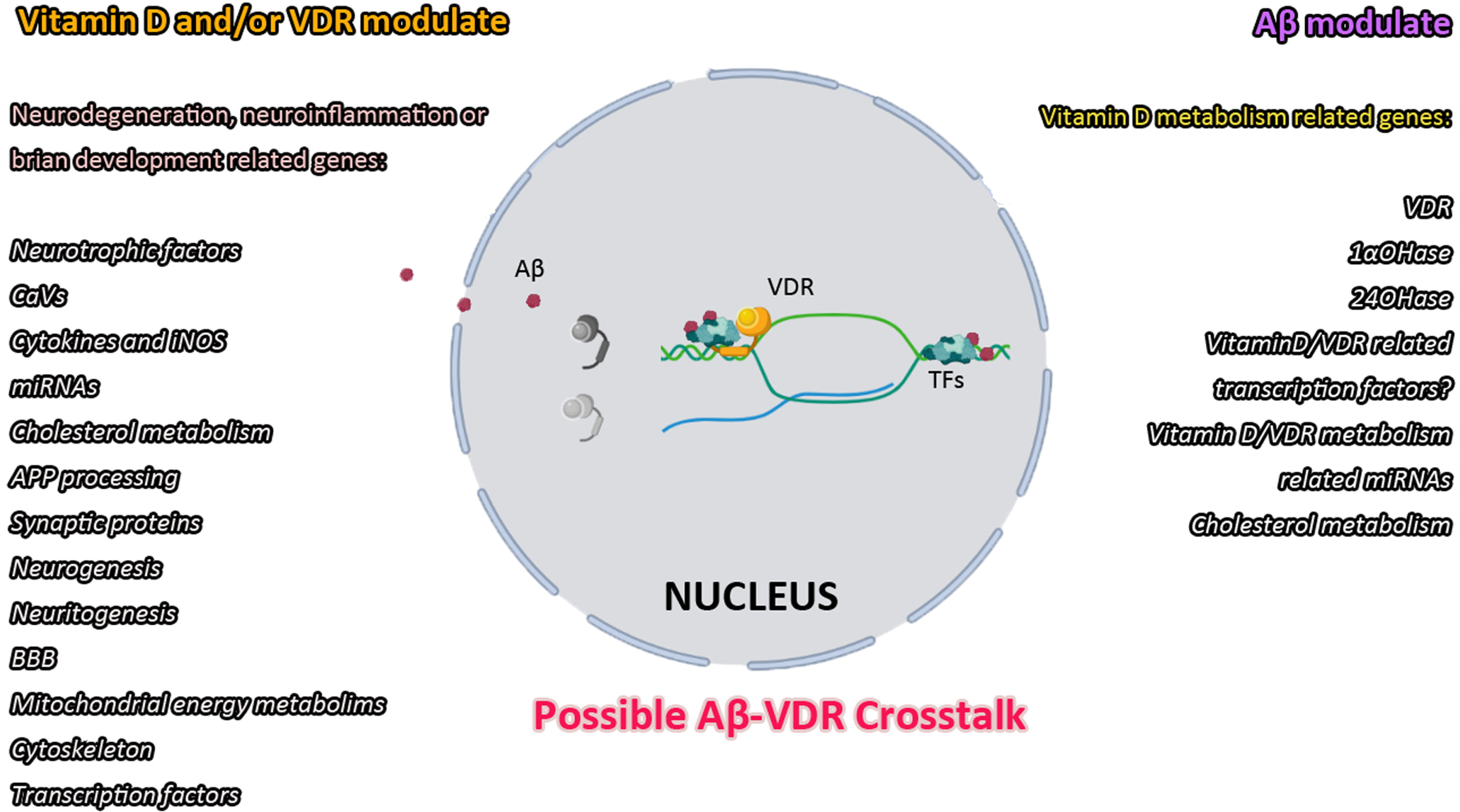
Aβ and vitamin D/VDR cross-talk: Studies indicated that vitamin D/VDR action is included in most neurodegeneration-related mechanisms. Aβ is also reported to alter vitamin D/VDR metabolism, leading to inefficient utilization of vitamin D and more vulnerable neurons.
A GENERAL PERSPECTIVE FOR VITAMIN D IN LATER STAGES OF LIFE
For a long time, we have known that VDR knockout mice had a shorter lifespan and presented premature aging [58]. However, researchers have not been interested much in the brains of these animals except for limited number of studies [58, 266]. Recently, a study conducted in the model organism Caenorhabditis elegans (C. elegans) showed that vitamin D significantly extended the lifespan and health span of wild-type worms too. Bioinformatic analysis of this study suggested vitamin D is associated with innate immune response and metabolism of xenobiotic compounds in the worms. They also treated the worms genetically modified to model AD with vitamin D, but in this case, vitamin D minimally extended the lifespan of these worms [267]. Our previous studies and others have provided enough background for such an effect. We suggested that the consequences of long-term vitamin D deficiency, i.e., hypovitaminosis D and/or inefficient utilization of vitamin D, may cause neurons to be vulnerable to aging and neurodegeneration [5–8]. In addition, we have proposed AD as an outcome of long-term hormonal imbalance, the crucial hormone vitamin D [8–12]. This suggestion may be supported by Kivimäki et al. since they determined high body mass index, which was possibly correlated with low vitamin D levels, 30 years before disease symptoms and progression to low body mass index, which may indicate high energy consumption and higher oxidative stress, during the preclinical stages of AD [268]. Even Braak et al. interpreted sporadic AD as being the result of a tauopathy, possibly beginning in childhood; and the effects of amyloid after a given threshold is crossed [269]. Vitamin D, 80% of which is produced from the skin, may be working as a counter that determines the lifespan of the organism. If we consider vitamin D as an hourglass, the decrease in vitamin D production as we age causes the clock to expire. The premature aging of VDR knockout animals [58] is one of the best examples. For this reason, with the weakening of other mechanisms in the organism as we age [195], the decrease in the production and use of this molecule, which is synthesized by sun rays, may be one of the triggers of neurodegeneration. However, neurodegeneration should be looked at from a different perspective. The possible relationship of Aβ, which is primarily involved in the pathology of AD, with VDR and other proteins involved in vitamin D metabolism, may render it unable to function through enzymes that change the level of VDR and increase vitamin D catabolism, even if vitamin D is at a sufficient level. This situation requires us to look at vitamin D metabolism from a different perspective, especially in neurodegeneration, and to consider the possibilities of pathological components of neurodegenerative diseases to affect this metabolism. Maintaining a certain level of vitamin D after the onset of the disease may limit the destruction caused by pathological components and save time, but it does not seem likely to expect a complete recovery before the main cause of the pathology is eliminated. In this case, while it is important to keep vitamin D levels at an adequate levels at all stages of life [10], it seems more appropriate to consider new approaches such as agonists that can activate VDR activity as well as additional supplements after the disease. However, it should be kept in mind that oral supplements will never produce other derivatives produced in the synthesis process with UVB.
ASSESSMENT OF THE ASSOCIATION
Sir Austin Bradford Hill, a professor emeritus of medical statistics at University of London asked a question in his article: The Environment and Disease: Association or Causation? published in 1965 [270]. He asked this question to assess whether a factor can be the cause of a disease, fairly and objectively. Before deciding the causation, he indicated that nine aspects of the association we should especially consider. These were 1) the strength of the association, 2) consistency of the observed association, 3) specificity of the association, 4) temporal relationship of the association, 5) biological gradient (dose response) 6) plausibility, 7) coherence, 8) experiment, and 9) analogy [270]. cSuch evaluation was given below and summarized in Table 1. Assessing with Hill’s criteria indicated that the association between vitamin D/vitamin D deficiency/vitamin D-VDR axis and AD is significant and the association is strong. The association is consistently confirmed in different populations, different models, and experiments. Although not 100% confirmed in human studies, regardless of the species or gender of animal or even the type of model (transgenic or chemical/peptide induced) vitamin D treatment was beneficial in animal models of AD [5, 271–286]. Temporal relation is not 100% overruled. AD type pathology was reported to disrupt vitamin D synthesis, increase catabolism of vitamin D, and suppress VDR expression. Besides, the disruption of vitamin D related receptors resulted in increased AD type pathology [7, 287]. VDR found to be as a mitochondrial transcription factor both in vitro and in human brain samples [241]. Only one study reported that vitamin D inversely correlated with known biomarkers of neurodegeneration in human CSF samples which indicates the dose response [156]. Though the other studies that used vitamin D treatment did not evaluate the question. Studies including cell cultures and animal models indicate that biological background is evident for vitamin D deficiency or disruption of vitamin D/VDR mechanisms to cause AD type pathology. The cause and the effect interpretation seems not conflicting with the generally known facts of AD. Yet generally known facts ignore the fact that vitamin D is secosteroid hormone and its level is tightly regulated in human body. The statement “VDR is a major player in energy metabolism of neurons as a transcription factor” was not a part of literature until 2023. The relation between vitamin D levels and AD biomarkers was not a matter of concern until recently [7, 287]. Studies indicated that there is a high possibility of preventing AD pathology or related changes with suitable vitamin D supplementation [7, 272–287]. However, analogy for vitamin D/VDR axis in AD and another drug/hormone molecule or disease is almost impossible, given the fact that vitamin D is a secosteroid hormone which acts mostly via VDR a nuclear and mitochondrial transcription factor and a possible signal relaying receptor that regulates more than a thousand gene and major signaling pathways [162, 288–290].
Assessment of the association between vitamin D/vitamin D deficiency/VDR axis and Alzheimer Disease according to Hill’s criteria
RCT, randomized controlled trial; OS, observational studies; SR, systematic reviews; MA, meta-analyses; PS, pilot study; RA, review article; LS, longitudinal study; CCS, cell culture studies; AM, animal models.
CONCLUSION
Neurodegeneration and vitamin D deficiency are both a long-term process in a human life span. Given its involvement in many cellular mechanisms, especially in the maintenance of healthy neuronal cells and in the regulation of pathological hallmarks of AD, we defined AD as an outcome of long-term hormonal imbalance of vitamin D. Though as we studied this concept last decade, we and others concluded that some of these concepts regarding neurodegeneration, might depend on vitamin D, its metabolites, its receptors, and other proteins that are involved in vitamin D metabolism together or individually. Given that the vitamin D field covers all these aspects should be taken into account when researching vitamin D-neurodegeneration relation. Utilizing agonists that can activate vitamin D receptors, and other derivatives produced in the synthesis process with UVB are all crucial when considering vitamin D-based intervention studies. Since there is a cross-talk between Aβ and vitamin D/VDR axis we should also consider inefficient utilization of vitamin D as a fact in AD-type pathology. Thus keeping vitamin D at adequate levels at all stages of life to establish high cognitive reserve might be crucial.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The reviewed studies that are performed in Istanbul University-Cerrahpasa are supported by the Scientific and Technological Research Council of Turkey-TUBITAK (Project No. 115S438, 214S585, 214S586, 216S887, 217S375, 219Z179) and by Research Fund of Istanbul University-Cerrahpasa (Project No: 30666, 33479, 34211, 35917). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.