
Abstract
Background:
At least one-third of Alzheimer’s disease (AD) patients have cerebrovascular abnormalities, micro- and macro-infarctions, and ischemic white matter alterations. Stroke prognosis impacts AD development due to vascular disease. Hyperglycemia can readily produce vascular lesions and atherosclerosis, increasing the risk of cerebral ischemia. Our previous research has demonstrated that protein O-GlcNAcylation, a dynamic and reversible post-translational modification, provides protection against ischemic stroke. However, the role of O-GlcNAcylation in the exacerbation of cerebral ischemia injury due to hyperglycemia remains to be elucidated.
Objective:
In this study, we explored the role and underlying mechanism of protein O-GlcNAcylation in the exacerbation of cerebral ischemia injury caused by hyperglycemia.
Methods:
High glucose-cultured brain microvascular endothelial (bEnd3) cells were injured by oxygen-glucose deprivation. Cell viability was used as the assay result. Stroke outcomes and hemorrhagic transformation incidence were assessed in mice after middle cerebral artery occlusion under high glucose and streptozotocin-induced hyperglycemic conditions. Western blot estimated that O-GlcNAcylation influenced apoptosis levels in vitro and in vivo.
Results:
In in vitro analyses showed that Thiamet-G induces upregulation of protein O-GlcNAcylation, which attenuates oxygen-glucose deprivation/R-induce injury in bEnd3 cells cultured under normal glucose conditions, while aggravated it under high glucose conditions. In in vivo analyses, Thiamet-G exacerbated cerebral ischemic injury and induced hemorrhagic transformation, accompanied by increased apoptosis. While blocking protein O-GlcNAcylation with 6-diazo-5-oxo-L-norleucine alleviated cerebral injury of ischemic stroke in different hyperglycemic mice.
Conclusion:
Overall, our study highlights the crucial role of O-GlcNAcylation in exacerbating cerebral ischemia injury under conditions of hyperglycemia. O-GlcNAcylation could potentially serve as a therapeutic target for ischemic stroke associated with AD.
Keywords
INTRODUCTION
Ischemic stroke is a common cerebral disease that occurs when the blood supply to the brain is interrupted or reduced, preventing brain tissue from getting oxygen and nutrients. With the increasing aging of the world, ischemic stroke is becoming one of the most common serious diseases, with high incidence, death, and disability. It is a major medical problem that needs to be solved urgently [1, 2]. Recent research also revealed stroke increases the risk of dementia, including Alzheimer’s disease (AD) [3]. Patients with AD dementia also have a higher risk of ischemic stroke [4]. Recently, by using of mouse model of cerebral ischemia, we reported a mechanism by which lysosomal enzyme asparagine endopeptidase is associated with brain ischemia and tau hyperphosphorylation, which is the hallmark of AD [5]. Many retrospective clinical studies have shown that hyperglycemia is a significant independent risk factor for high incidence and mortality of stroke after excluding factors such as age, sex, blood pressure, and arterial disease. Hyperglycemia significantly contributes to poor prognosis in patients with acute ischemic stroke [6]. Hyperglycemia occurs more frequently in patients with diabetes, with a four times higher risk of ischemic stroke than the general population [7].
Additionally, diabetes significantly aggravates hemorrhagic brain injury and leads to higher mortality rates. At the same time, clinically, about 40–50% of ischemic stroke patients without diabetes are diagnosed with hyperglycemia upon hospital admission, which is called poststroke hyperglycemia (PSH). Hyperglycemia can affect the function of the vascular endothelium, exacerbate the risk of hemorrhagic transformation (HT) after cerebral ischemia, and increase the rate of intracranial hemorrhage after thrombolysis or mechanical thrombectomy. Animal experiments also confirmed that streptozotocin (STZ)-induced type I diabetes or hyperglycemia induced by glucose injection in mice resulted in more severe cerebral injury than in normal mice. Cerebral ischemia injury is exacerbated by hyperglycemia, possibly due to oxidative stress, inflammation, and reduced blood reperfusion after flow recovery, although this mechanism has not been widely accepted [8]. Understanding the post-ischemic cellular response is crucial for the recovery and survival of an organism and will help develop more effective strategies to treat ischemic brain injury.
O-GlcNAcylation is an essential post-translational modification of numerous proteins within the nucleus, cytoplasm, and mitochondria. It significantly impacts various biological processes, including transcription, signal transduction, and metabolism [9–11]. The dynamic addition and hydrolysis of N-acetylglucosamine (GlcNAc) to serine/threonine residues are catalyzed by a unique couple of enzymes: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). UDP-GlcNAc, the donor substrate for OGT, is the end-product of the nutrient-sensitive hexosamine biosynthesis pathway (HBP). HBP, a cellular trophic sensor, is linked to energy metabolism abnormalities in diabetes mellitus in numerous investigations. Since O-GlcNAcylation changes due to changes in metabolic flux through HBP, O-GlcNAc modification has been considered a “sensor” of energy and stress [12]. Dysregulation of O-GlcNAc is associated with complex diseases that severely threaten human health, including cancer, diabetes, and neurodegenerative diseases, including AD. We have revealed that cerebral glucose hypometabolism leads to abnormal hyperphosphorylation of tau via downregulation of tau O-GlcNAcylation in AD [13]. Our and other studies have found that the induction of protein O-GlcNAcylation protects against myocardial ischemia and cerebral ischemic injury in both young and old animals [14–16]. Pharmacological elevation of O-GlcNAcylation after ischemic stroke promotes cell survival, convincingly demonstrating the protective effect of O-GlcNAc [17, 18]. However, emerging evidence has shown that in diabetic cardiomyopathy, persistently high levels of O-GlcNAc contribute to a decrease in the regulation of insulin transduction signals and sensitivity to stress, diminishing the cardioprotective effects of insulin against ischemia/reperfusion injury and increasing the risk of vascular calcification, and overexpression of OGA to decrease O-GlcNAc modification can significantly improve cardiac function [19]. Treatment of retinal neurons with high glucose diminished the protective effect of insulin and induced apoptosis owing to an excessive increase in HBP flow compared to neurons cultured in normal glucose concentration [20]. Therefore, these data convincingly suggest that O-GlcNAcylation is an endogenous protective mechanism against stress. In contrast, excessive or persistent upregulation of O-GlcNAc levels may be directly linked to the adverse effects of hyperglycemia, including insulin resistance, glucose toxicity, and pancreatic β-cell death.
The development of HT after ischemia is related to the destruction of the blood-brain barrier (BBB). The BBB is a vital defense structure of the central nervous system (CNS) that can restrict the entry of water-soluble and macromolecular lipid-soluble substances into the brain. It has been reported that high glucose induces apoptosis in brain microvascular endothelial cells [21]. Diabetes characterized by hyperglycemia is prone to induce HT after cerebral ischemia injury owing to microvascular damage and consequently disruption of the BBB [22]. Our previous study showed that an excessive increase in O-GlcNAcylation induces neuronal apoptosis in vivo under cerebral ischemia-reperfusion [23]. To determine the contribution of O-GlcNAcylation to cerebral ischemic injury under hyperglycemia, we used various animal and cell models of diabetes and PSH. The level of intracellular O-GlcNAc is regulated pharmacologically. We explored the molecular mechanism through which O-GlcNAcylation induces cell apoptosis in hyperglycemia combined with stroke.
MATERIALS AND METHODS
In vitro culture of cells
The mouse brain endothelial cell line bEnd.3 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). DMEM (Low-glucose Dulbecco’s modified eagle medium) with 4% L-glutamine, 1.5 g/L sodium bicarbonate, and 1 g/L glucose (Sangon Biotech, Shanghai, China) containing 10% fetal bovine serum and 10% penicillin/streptomycin (Gibco, Grand Island, NY, USA) was used to culture the cells. Moreover, the cells were incubated in a humidified environment at 37°C with 5% CO2 used for subsequent experiments 24 h after cell assay plating.
In vitro model of oxygen deprivation and reoxygenation
To simulate OGD/R stress in vitro, the cell medium was removed and bEnd.3 cells were rinsed several times with glucose-free DMEM (Gibco). The cells were transferred to a glucose-free DMEM medium in a box filled with 95% N2 and 5% CO2 for 6 h. Afterward, the glucose-free DMEM was removed, and bEnd.3 cells were grown in complete DMEM containing 10% FBS for 18 h, thus simulating reperfusion. The control group cells were treated identically to the experimental group, except that they did not receive OGD/R (Fig. 1b).
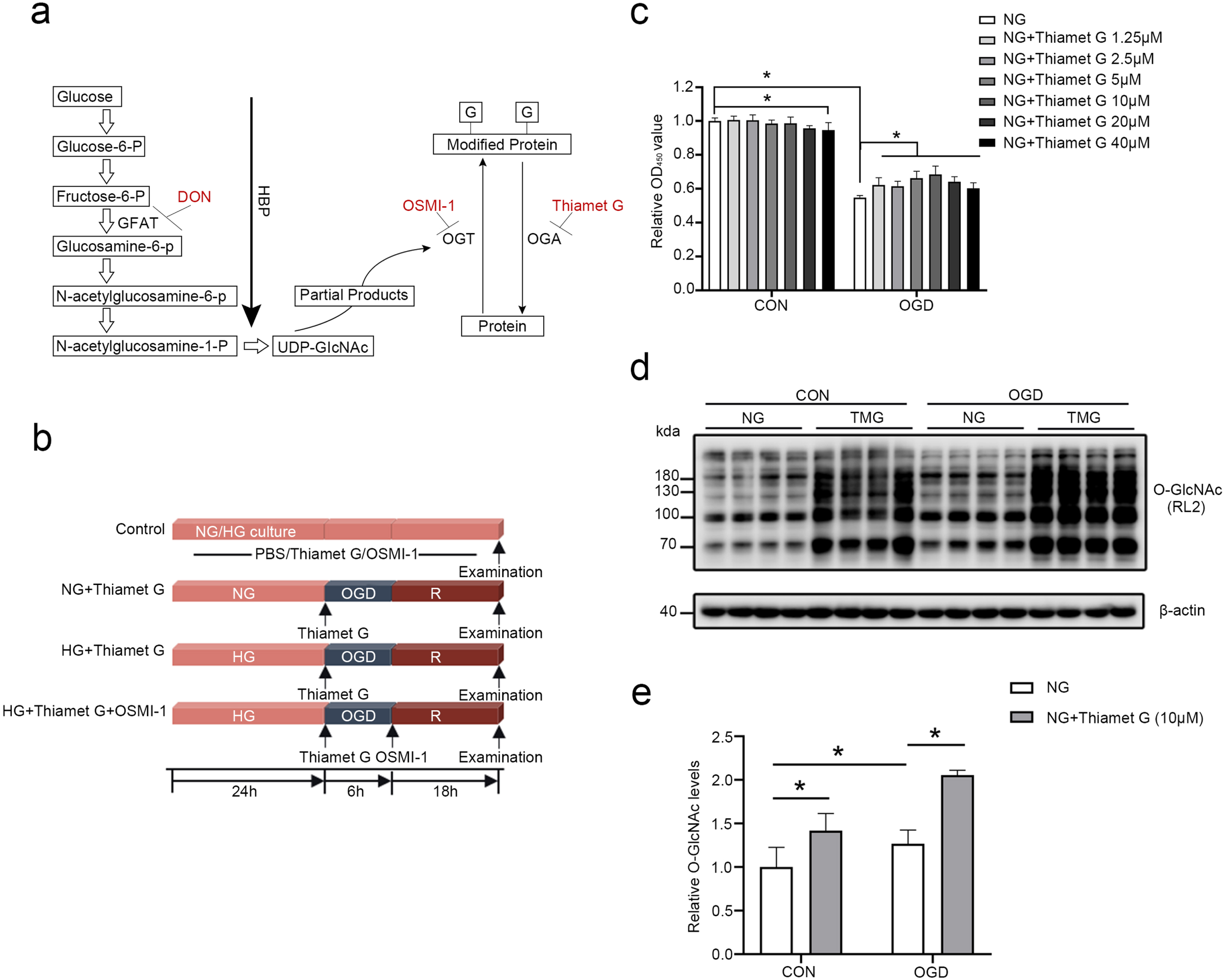
TMG induces upregulation of the protein O-GlcNAcylation, which attenuates OGD/R-induced injury in bEnd3 cells cultured under normal glucose conditions. (a) Diagram of HBP and protein O-GlcNAcylation. TMG, DON, and OSMI-1 raised or decreased (red) O-GlcNAcylation levels. (b) Construction of an OGD/R cell model and experimental design diagram. bEnd3 cells were cultured in normal glucose (NG, 5.56 mM glucose) or high glucose for 24 h. OGD was performed for 6 h, followed by 18 h of reoxygenation (OGD/R). TMG was added at the beginning of OGD, and OSMI-1 was added at the beginning of reoxygenation. The control group (CON) was maintained in an NG culture medium under a normoxic atmosphere and treated with phosphate buffer (PBS). (c) Histogram illustrating the cell viability of control cells, cells subjected to 6 h of OGD and 18 h of reoxygenation alone, and cells treated with different concentrations of TMG (1.25μM, 2.5μM, 5μM, 10μM, 20μM, 40μM) throughout the OGD/R phase. (d-e) Western blot analysis of O-GlcNAcylation in cells treated with 10μM TMG. All the quantification data are presented as the mean±SD (n = 4). *p < 0.05.
Drug administration and cell viability measurement in vitro
The viability of bEnd.3 cells were assessed by the Cell Counting Kit (CCK-8) cytotoxicity assay (Dojindo Laboratories, Kumamoto, Japan). Briefly, bEnd.3 cells (1×104 cells/well) were seeded in a 96-well plate in DMEM. Normal bEnd3 cells or bEnd3 cells subjected to OGD/R were treated with different concentrations of TMG or OSMI-1. TMG was added at the beginning of OGD, and OSMI-1 was added at the beginning of reoxygenation. The experiments were completed by adding 10μL of CCK-8 reagent to each well and incubating it for 1 h in the dark at 37°C. The absorbance at 450 nm was measured using an automatic microplate reader (Bio-Tek, Winooski, VT, USA). A sextuple experiment was conducted in triplicates.
Research animals and biological reagents
C57BL/6 male mice weighing 20–22 g were purchased from Vitalriver (Beijing, China) at five weeks of age. The mice were housed in six cages at the Institute of Laboratory Animal Center at Nantong University under standard conditions with 12 h light/dark cycles. All animal procedures were reviewed and approved by the Nantong University Animal Care Committee committee with the approval number of IACUC20220201-1010 and experimental procedures were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
TMG (MedChemExpress, Princeton, NJ, USA), 6-diazo-5-oxo-L-norleucine (DON), and OSMI-1 (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in phosphate-buffered saline (PBS). STZ and 2,3,5-triphenyltetrazolium chloride (TTC) were obtained from Sigma-Aldrich. We used antibodies against RL2 (Thermo Fisher, Waltham, MA, USA), Cleaved Caspase 3, Bcl-2, and Bax (Cell Signaling Technology, Beverly, CA, USA), and β-actin (Sigma-Aldrich, St. Louis, MO, USA).
Establishment of different hyperglycemic mouse models with cerebral ischemic/reperfusion (I/R) injury
STZ-induced chronic hyperglycemia model
The mice were acclimated to a well-ventilated room. We intraperitoneally injected mice with 45 mg/kg STZ in 0.1 mol/L sodium citrate buffer (pH 4.3 4.5) for five consecutive days to induce diabetes. FreeStyle Optium Neo meters (Yuwell, Zhenjiang, China) were used to measure blood glucose levels after STZ treatment. The mice used for the experiment had fasting blood glucose values greater than 16.7 mmol/L after STZ injection. Throughout the modeling process, the health status of mice was consistently observed, and none of the mice died. Each mouse’s weight, food intake, and water consumption were also recorded.
Four weeks after STZ injection, the diabetes mellitus mice (DM) were randomly divided into four groups: (a) diabetic sham-operated mice (DM-sham group); (b) diabetic I/R injured mice (DM-middle cerebral artery occlusion (MCAO) group); (c) diabetic I/R injured mice treated with TMG 30 mg/kg (DM-MCAO+TMG group); and (d) diabetic I/R injured patients treated with 2.6μg of DON (DM-MCAO+DON group). A 30 mg/kg dose of TMG, an O-GlcNAcase-specific inhibitor, was administered intraperitoneally 3 h before MCAO. The concentration of TMG was determined based on a previous study [18]. In diabetic mice, thread emboli were injected into the right middle cerebral artery (MCA) to induce cerebral I/R injury. A gentle withdrawal of the filament, followed by 24 h of reperfusion, was performed after 60 min of blocking.
High glucose-induced post-ischemic acute hyperglycemia model
The mice were acclimated to a well-ventilated room. The high glucose (HG) induced mice were randomly divided into four groups: (1) HG-sham, (2) HG-MCAO, (3) HG-MCAO+TMG, and (4) HG-MCAO+DON. TMG was intraperitoneally injected at 30 mg/kg 3 h before MCAO. A dose of 6 ml/kg of 50% glucose was injected intraperitoneally 5 min before MCAO. FreeStyle Optium Neo meters (Yuwell, Zhenjiang, China) measured blood glucose levels after MCAO at four time points. After 75 min of blocking, the filament was gently withdrawn, and reperfusion was allowed for 2 h.
MCAO
Mice were heavily anesthetized with isoflurane gas at a rate of 5% isoflurane and maintained at 3.5% isoflurane throughout surgery (RWD, Shenzhen, China). An intraluminal filament was inserted through the external carotid artery stump (ECA) into the internal carotid artery (ICA) and gently advanced 11 mm beyond the carotid bifurcation to occlude the MCA. Cerebral blood flow (CBF) was monitored using laser speckle contrast imaging (RFLSI III, RWD), and animals presenting with unsuccessful blood flow obstruction or reperfusion in CBF were excluded from further study. After blocking for 60 min (DM mice) or 75 min (HG mice), the filaments were gently removed, and reperfusion was allowed for 24 h. Blinded to the experimental design, an experienced investigator conducted MCAO. The body temperature of the mice was kept at 36.5–37.5°C during the operation using a heat lamp. Mice in the sham-operated group underwent similar procedures, apart from filament insertion.
Behavioral tests
Blinded investigators measured focal and general neurological deficits 24 h after reperfusion [24].
Focal neurological deficits were measured to evaluate symmetry, gait, climbing, circling, and sensory responses after MCAO. General neurological deficits were measured to assess hair, ears, eyes, posture, spontaneous activity, and epileptic behavior after MCAO.
The final score was calculated as the mean of three repeated trials. Higher scores indicate more severe neurological impairment. A team of two independent researchers analyzed the scores.
TTC staining
TTC staining was performed to determine the size of the cerebral infarct. The infarct size was calculated as the ratio of infarct area to total area. Brains were cut into 2 mm thick coronal sections. The sections were soaked in a 1% TTC solution for 30 min at 37°C, stained while turning over, and fixed overnight in a 4% paraformaldehyde solution. The slices were neatly arranged and scanned in layers (CanoScan LiDE 300 CN; Canon INC, Beijing, China). A blinded observer determined the infarct volume and corrected it for edema using ImageJ (version 1.8.0; National Institutes of Health, Bethesda, MD, USA).
Intracerebroventricular injection
We anesthetized mice with 2.5% avertin and then restrained them onto a stereotaxic apparatus. The bregma coordinates used for injection was: –1.0 mm lateral, –0.3 mm posterior, and –2.5 mm below. Each mouse received a single intracerebroventricular (icv) injection of DON dissolved in 4.0μL 0.9% saline or, as a control, 4.0μL 0.9% saline alone into the right ventricle of the brain. After slowly administering the injection over 5 min, the needle was retained for another 3 min before being removed.
Histological analysis of hemorrhagic transformation
To assess hemorrhagic changes, the brains were carefully removed 24 h after reperfusion and sliced into consecutive cross-sections of 2-mm thickness. Based on a previous study [25], hemorrhagic transformations can be classified into five categories: (0) non-hemorrhage; (1) small petechiae typically lining the boundary of an infarct, characteristic of hemorrhagic infarction type 1; (2) more confluent petechiae in the damaged area than in type 1 hemorrhagic infarction; (3) type 1 parenchymal hemorrhage characterized by blood clots < 30 percent of the injured parenchyma; (4) type 2 parenchymal hemorrhage characterized by blood clots > 30 percent of the injured parenchyma. Hemorrhagic scores were calculated as 0–4.
Hemoglobin assay
At 24 h after reperfusion, mice were injected intraperitoneally with 2.5% avertin (2,2,2-tribromoethanol, Sigma-Aldrich) at 400 mg/kg and perfused with room temperature saline transcardially. Briefly, the brains were removed and cut into four thick coronal sections. The contralateral (normal) and ipsilateral (infarct) sides were separated in each section. HT was quantified by measuring hemoglobin content in the brain.
*Accounts for sample dilution in the wells.
The tissue was homogenized in 5–10 ml of PBS solution, pH 7.4, containing 0.16 mg/ml heparin per gram weight of tissue, and centrifuged at 10,000×g for 15 min at 4°C. The supernatants were collected and stored on ice. The final volume of the assay was 200μL (20μL of sample and 180μL of Hemoglobin Detector) in all wells (Hemoglobin Colorimetric Assay Kit, Cayman, Ellsworth, MI, USA). The plate was covered with a plate cover and incubated at room temperature for 15 min. An automatic microplate reader (Bio-Tek, Winooski, VT, USA) was used to measure the absorbance at 560–590 nm.
Western blot
Western blotting was performed following previously described protocols [26]. Homogenized ischemic brain tissues were centrifuged, and supernatants were collected. BCA assays (Beyotime, Shanghai, China) were used to determine total protein content as described by the manufacturer. An equal amount of protein extract was resolved using 10–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto Immobilon-P membranes (Millipore, Bedford, MA, USA). Non-specific sites on the membranes were blocked in blocking buffer [Nonfat dry milk (5%) in 1×TBST (Tris-buffered saline containing 1% Tween-20)] for 60 min and incubated with the primary antibodies in Quick Blocktrademark Primary Antibody Dilution Buffer for western Blot (Beyotime, Shanghai, China) overnight at 4°C. The primary antibodies used were as follows: RL2 (1 : 1000, Thermo Fisher, Waltham, MA, USA), cleaved caspase 3 (1 : 500, Cell Signaling Technology (CST), Beverly, CA, USA), Bax (1 : 1000, CST), Bcl-2 (1 : 1000, CST), and β-actin (1 : 5000, Sigma-Aldrich, St. Louis, MO, USA). Next, horseradish peroxidase-conjugated goat anti-mouse IgG (H + L) (1 : 3000) or horseradish-conjugated goat anti-rabbit IgG (H + L) (affinity purified) (1 : 3000) was incubated with the membranes for 2 h at room temperature. Immunoreactive bands were visualized using an Odyssey imaging system (LI-COR Biosciences, Lincoln, NE, USA) and enhanced chemiluminescence (ECL; Bio-Rad, Hercules, CA, USA). The semi-quantification of bands was performed by densitometry using ImageJ software.
TUNEL assay
Apoptosis was detected using the TUNEL BrightGreen Apoptosis Detection Kit (Vazyme, Nanjing, China), according to the manufacturer’s instructions. Immediately after the indicated treatments, bEnd3 cells were fixed with 4% formaldehyde for 25 min at room temperature, permeabilized with 0.2% Triton X-100 for 5 min at room temperature, and then incubated with 50 mL of TdT enzyme buffer for 1 h at 37°C. A fluorescence microscope (PCM 200; Nikon, Tokyo, Japan) was used to observe the cells after counterstaining with DAPI for 5 min.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 8.0.1 software (GraphPad Software, San Diego, CA, USA). As a first step, Kolmogorov-Smirnov tests were conducted on all data to determine whether they were Gaussian. Two-way ANOVA was used to analyze the in vitro data when appropriate, and ANOVA was used to analyze the in vivo from the mouse experiments. Statistical significance was set at a probability value < 0.05.
RESULTS
TMG induces upregulation of protein O-GlcNAcylation, which attenuates OGD/R-induce injury in bEnd3 cells cultured under normal glucose conditions
The HBP is a glucose metabolic pathway. Normally, only 2% to 5% of glucose enters the HBP. The rate-limiting enzyme, glutamine: fructose-6-P amidotransferase (GFAT), enhances HBP flux to generate uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), the substrate for OGT. OGT uses UDP-GlcNAc to transfer O-linked β-N-acetylglucosamine (O-GlcNAc) moiety at serine or threonine residues of target proteins. OGA removes O-GlcNAc from modified proteins (Fig. 1a).
To explore the role of O-GlcNAcylation in acute brain ischemic injury, we used bEnd3 cells to establish an in vitro experimental hypoxia-reperfusion injury model. As shown in Fig. 1b, bEnd3 cells cultured in normal glucose (NG) or different concentrations of high glucose (HG) for 24 h were exposed to hypoxia for 6 h and reoxygenated for 18 h to induce OGD/R injury. To investigate the role of O-GlcNAcylation in the effects of the combination of hyperglycemia with cerebral ischemia, normal bEnd3 cells or bEnd3 cells subjected to OGD/R were treated with different concentrations of TMG (a specific inhibitor of OGA) or OSMI-1 (a cell-permeable OGT inhibitor. The cell viability was measured to determine if TMG is cytotoxic under normal conditions. TMG did not exhibit toxicity up to 10μM, although it caused a dose-dependent reduction in cell viability from 10 to 40μM, indicating that 10μM TMG is safe. The impact of TMG on cell survival after OGD/R insult was investigated. OGD/R significantly decreased cell viability; however, TMG treatment was highly resistant to OGD/R-induced decrease in the viability of bEnd3 cells. Furthermore, the effect of TMG (1.25–10μM) displayed a concentration-dependent pattern, and 10μM of TMG showed the most substantial protective effects in bEnd3 cells subjected to OGD/R (Fig. 1c). Therefore, 10μM TMG was selected in subsequent experiments. Next, we identified the intracellular O-GlcNAcylation levels induced by TMG. The results showed that O-GlcNAcylation levels were upregulated in both normal and OGD/R-injured cells after treatment with TMG (Fig. 1d, e). These data indicate that TMG dramatically increased intracellular O-GlcNAcylation to improve cell survival under normal glucose conditions.
Upregulation of O-GlcNAcylation levels aggravates OGD/R-induced injury in bEnd3 cells cultured under HG conditions
Next, we investigated the function of O-GlcNAcylation in bEnd3 cells grown in HG under normal and OGD/R stress conditions. According to the CCK8 assay, OGD/R induced a significant decrease in the viability of bEnd3 cells compared to that of the control. Moreover, HG (25, 40, 60, 80 mM) significantly exacerbated the decline in the viability of bEnd3 cells in a dose-dependent pattern (Fig. 2a). After HG stimulation, TMG induced a significant decrease in the viability of normal bEnd3 cells or bEnd3 cells subjected to OGD/R (Fig. 2b). However, the reduction in the viability of bEnd3 cells induced by TMG was entirely abolished by the OGT inhibitor OSMI-1 (Fig. 2c). Similarly, treatment of HG-stimulated cells with Thiamet G resulted in a significant increase in intracellular O-GlcNAcylation levels strikingly inhibited by 10μM OSMI-1 (Fig. 2d–g). These results demonstrate that upregulation of O-GlcNAcylation by TMG exacerbates OGD/R-induced cell damage in bEnd3 cells cultured in HG.
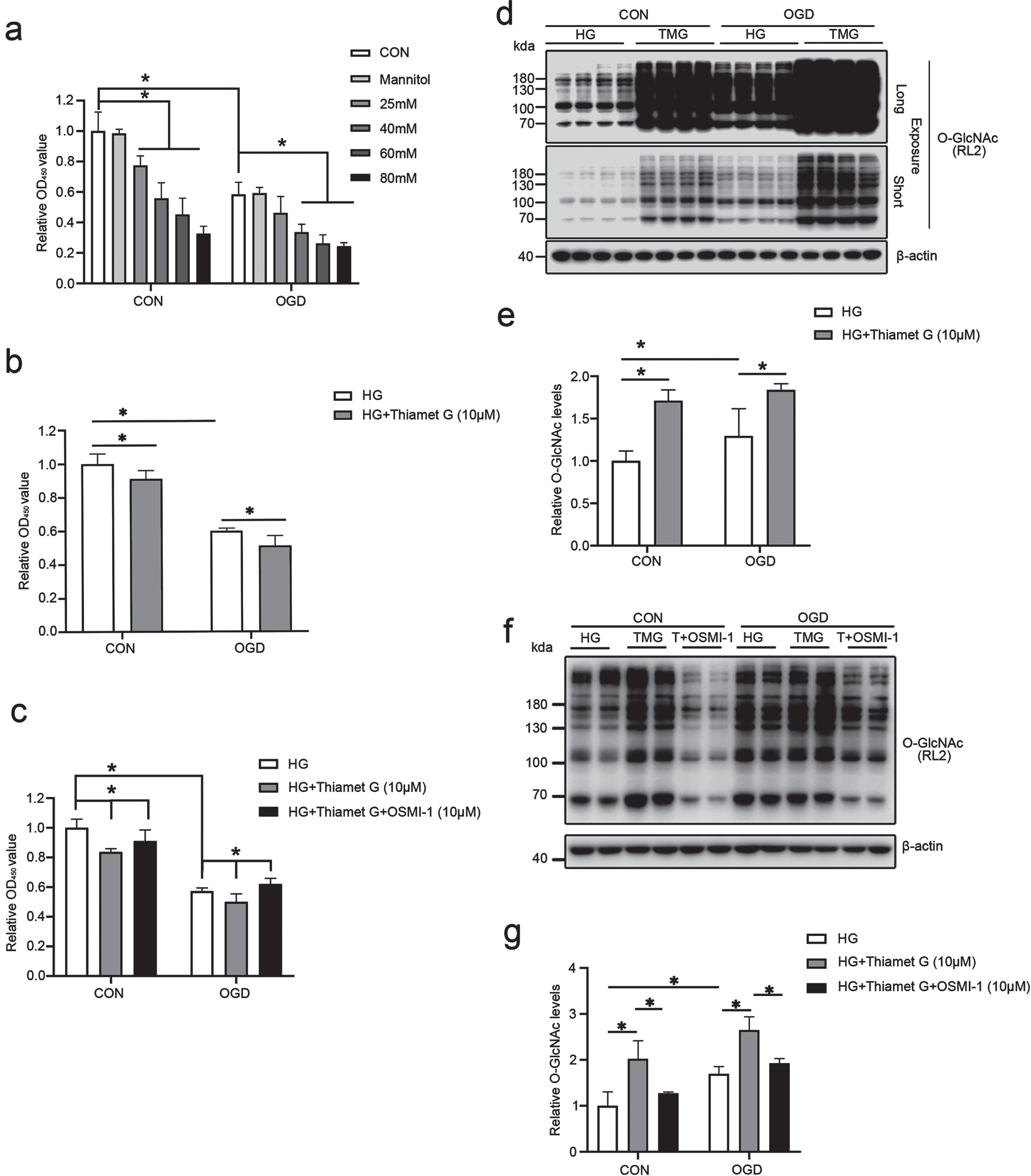
Upregulation of O-GlcNAcylation level aggravates OGD/R-induced injury in bEnd3 cells cultured under HG conditions. (a) Histogram depicting the viability of bEnd3 cells cultured in different concentrations of glucose (25, 40, 60, 80 mM glucose). Mannitol was used as osmolality control. (b) Histogram illustrating the viability of control and HG-stimulated cells (40 mM), cells exposed to OGD stress, and cells treated with 10μM TMG during the OGD/R period. (c) Cells were treated with HG (40 mM), 10μM TMG or TMG in combination with OSMI-1 (10 M) before the CCK-8 test was performed to determine the viability of bEnd3 cells. (d-g) Immunoblotting of protein O-GlcNAcylation using the RL2 antibody, followed by quantification of each band intensity adjusted to β-actin. Short-term and long-term exposure of western blots. All the quantification data are presented as the mean±SD (n = 3–4). *p < 0.05.
TMG aggravates cerebral infarction of cerebral ischemia in chronic hyperglycemic mice
To further verify the role of O-GlcNAcylation in hyperglycemia exacerbated cerebral ischemic injury in vivo, we established a diabetic model combined with cerebral ischemia-reperfusion. The in vivo experimental design was as follows: five-week-old C57BL/6 mice were acclimatized for several days and intraperitoneally injected with 1% STZ at a dose of 45 mg/kg for five consecutive days, followed by 2 weeks of continuous feeding and regular observation of body weight, drinking water consumption, and food consumption. Blood glucose levels were measured after 6 h of fasting. After confirming the success of the model, diabetic mice were divided into control (diabetic mice, DM), ischemic stroke (DM-MCAO), and ischemic stroke combined with TMG (DM-MCAO+TMG) groups, followed by pathological feature and biochemical examinations to assess neurological deficits 24 h after MCAO (Fig. 3a). The mice were kept for 2 weeks, but it is possible that the feeding time was short, and diabetic mice tended to lose weight compared to normal mice, but no difference was observed (Fig. 3b). In diabetic mice, drinking water and food intake were significantly increased with time (Fig. 3c, d). Compared with normal mice, fasting blood glucose values were consistently and significantly increased in diabetic mice (Fig. 3e). The laser scattergrams show cerebral blood flow was reduced after ischemia, and partial blood flow was restored to the brain after reperfusion (Fig. 3f). The above data indicate that diabetes combined with the cerebral ischemia-reperfusion model was successfully induced.
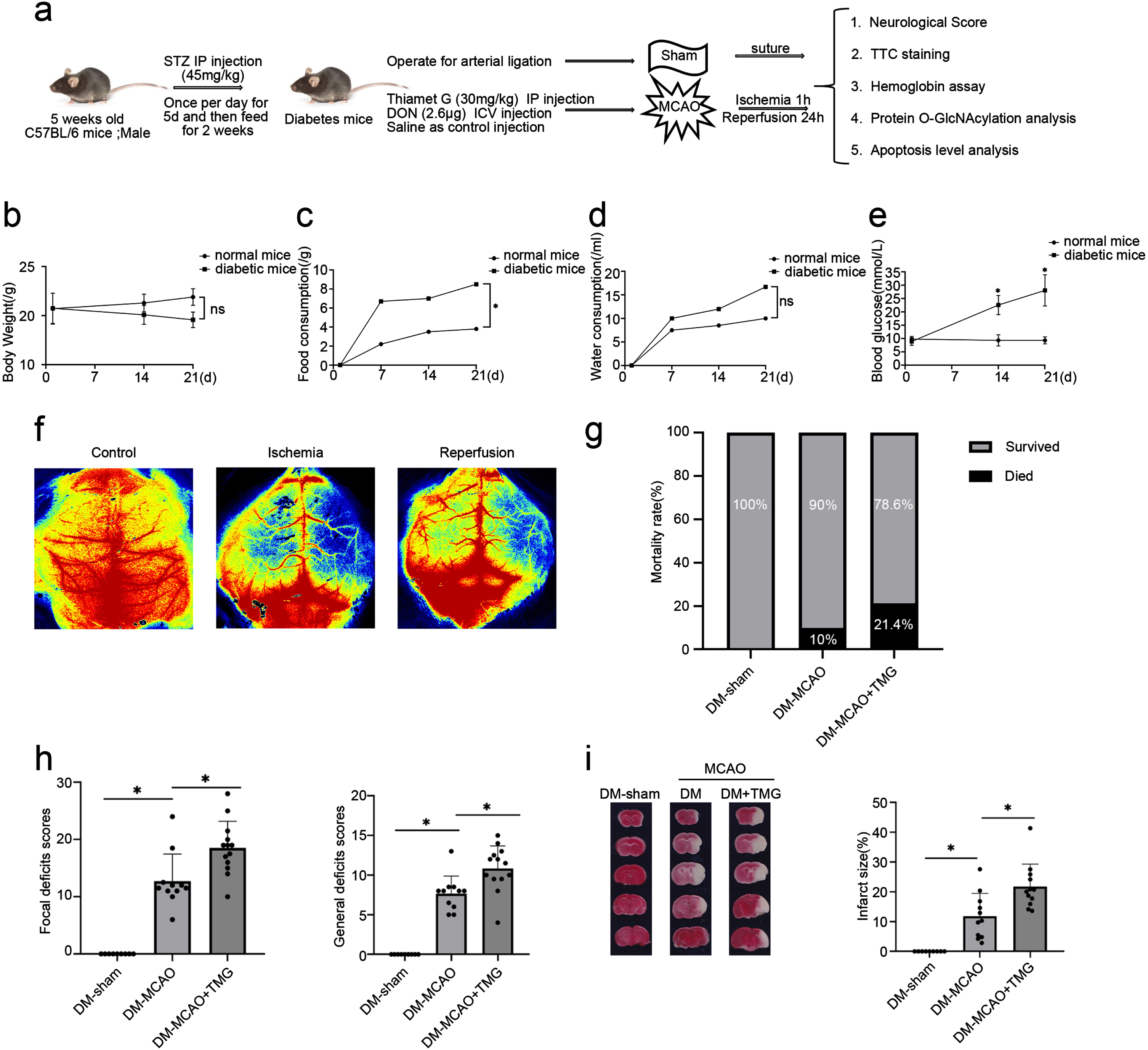
TMG aggravates cerebral infarction of cerebral ischemia in chronic hyperglycemic mice. (a) Schematic experimental design of drug treatments in a diabetic model subjected to cerebral ischemia-reperfusion injury. (b–e) Line charts show the changes in body weight, food consumption, and water consumption following STZ treatment. Blood glucose levels in diabetic mice were monitored for two weeks. (f) Laser scattergram to monitor cerebral blood flow. (g) The graph depicts the death rate 24 h following MCAO. (h) Neurological scores in DM-Sham, DM-MCAO, and DM-MCAO+TMG groups. (i) Photographs of coronal slices stained with TTC and measurements of brain infarct volume at 24 h after MCAO. Data are presented as mean±SD, *p < 0.05. DM-sham (n = 9), DM-MCAO (n = 11), and DM-MCAO+TMG groups (n = 13). STZ, streptozotocin; MCAO, transient middle cerebral artery occlusion; DM-sham, diabetes mellitus sham control; DM-MCAO, MCAO performed in diabetic mice treated with saline; DM-MCAO+TMG, DM-MCAO animals treated with TMG.
We previously reported that the upregulation of O-GlcNAcylation is neuroprotective following cerebral ischemia-reperfusion disease without hyperglycemia. In the present investigation, we aimed to determine whether increasing O-GlcNAcylation during hyperglycemia, which can be achieved by TMG, exacerbates ischemic stroke. Thus, we intraperitoneally injected TMG 3 h before ischemic stroke into diabetic mice. No significant difference in mortality was found among the diabetic mice in the three groups (Fig. 3g). However, neurological scores and the area of cerebral infarction in DM-MCAO mice remarkably increased compared to DM mice, and in DM-MCAO+TMG mice further exacerbated brain injury worsening neural function and expanding the size of cerebral infarction compared with DM-MCAO mice (Fig. 3h, i). These data suggest that TMG aggravates cerebral ischemic injury in chronic hyperglycemic mice.
TMG aggravates cerebral infarction of cerebral ischemia in post-ischemic acute hyperglycemic mice
In addition, we established an acute hyperglycemia model following cerebral ischemia-reperfusion to simulate clinical PSH. The in vivo experimental design was as follows: seven-week-old C57BL/6 mice were acclimatized for one week. Mice were divided into HG-sham, HG-MCAO, and HG-MCAO combined with TMG (HG-MCAO+TMG) groups. TMG was administered intraperitoneally at 30 mg/kg 3 h before MCAO. 50% glucose (6 ml/kg) was intraperitoneally injected 5 min before mice were subjected to ischemic surgery, followed by an assessment of neurological deficits (Fig. 4a). Blood glucose was measured at four time points: 0, 15, 45, and 90 min after glucose injection. Fifteen minutes after glucose injection, the blood glucose levels in the three groups rapidly increased to 30 mmol/L and then dropped to 20 mmol/L after 90 min, indicating that hyperglycemia was successfully induced (Fig. 4b). Similar to the results in chronic hyperglycemic ischemic mice, compared with the HG-sham mice, the mortality showed an increasing trend in HG-MCAO mice and further increased in HG-MCAO+TMG mice (Fig. 4c). Twenty-four hours after stroke, HG-MCAO mice exhibited significantly worse neurological scores and larger infarct volumes than HG-sham mice (Fig. 4d). It is worth noting that compared to HG-MCAO mice, TMG-treated mice showed worse stroke outcomes in neurologic scoring. In addition, the stroke infarct size was augmented in HG+TMG mice (Fig. 4e). These results, in conjunction with the poorer stroke prognosis previously observed in diabetic mice, suggest that further upregulation of O-GlcNAcylation under HG conditions is detrimental during the acute ischemic stroke phase.

TMG aggravates cerebral infarction of cerebral ischemia in post-ischemic acute hyperglycemic mice. (a) Schematic experimental design of drug treatments in the PSH model. (b) Line charts show the changes in blood glucose levels (Four time points, 0 min, 15 min, 45 min, and 90 min after 50% glucose injection). (c) Graph summarizes the mortality rate at 24 h after MCAO. (d) Neurological scores in HG-sham, HG-MCAO, and HG-MCAO+TMG groups. (e) Photographs of coronal slices stained with TTC and measurements of brain infarct volume at 24 h after MCAO. Data are presented as mean±SD, *p < 0.05. HG-sham (n = 9), HG-MCAO (n = 15), and HG-MCAO+TMG groups (n = 11). MCAO, transient middle cerebral artery occlusion; HG-sham, sham mice treated with 50% glucose; HG-MCAO, MCAO performed in mice treated with 50% glucose; HG-MCAO+TMG, HG-MCAO mice treated with TMG.
TMG aggravates cerebral ischemia prognosis and induces hemorrhagic transformation in different types of hyperglycemic mice
Increasing evidence indicates that BBB disruption is a hallmark of ischemic stroke, contributing to HT and poor prognosis [27]. The occurrence and severity of HT were assessed by determining the hemorrhagic scores in brain slices and measuring the hemoglobin content in the ischemic brain. Twenty-four hours after MCAO, diabetic mice had modest HT symptoms and a small bleeding region. TMG administration dramatically increased the incidence and severity of HT in the ischemic brains of diabetic mice (Fig. 5a). More obvious bleeding was observed in acute hyperglycemic mice after MCAO, and TMG further aggravated HT in ischemic brain tissue (Fig. 5b). We quantified the severity of HT according to the scoring criteria shown in Fig. 5c. The results showed that TMG dramatically increased HT in hyperglycemic animals with cerebral ischemia (Fig. 5d, e). Next, the hemoglobin content of different brains was determined. MCAO significantly increased hemoglobin content in the brains of diabetic mice. TMG injection further increased hemoglobin content in the ischemic brain tissue of diabetic mice (Fig. 5f). As shown in Fig. 5g, hemoglobin content in the HG-MCAO group showed a sharp rise compared with that in the HG-sham group, and TMG further increased the hemoglobin content. This evidence suggests that upregulation of O-GlcNAcylation by TMG aggravates HT after ischemic stroke under hyperglycemic conditions.

TMG aggravates cerebral ischemia prognosis and induces HT in different hyperglycemic mice. (a, d) Representative photos of HT at 24 h after MCAO. (b, e) Hemorrhagic scores of brain slices, according to the degree of bleeding. Data are reported as scores of animals showing signs of HT. (c, f, g) The hemoglobin content of different types of mice. Data are presented as mean±SD, *p < 0.05. DM-sham (n = 4), DM-MCAO (n = 6), and DM-MCAO+TMG groups (n = 7); HG-sham (n = 5), HG-MCAO (n = 15) and HG-MCAO+TMG groups (n = 16).
GFAT inhibitor DON alleviates cerebral infarction of cerebral ischemia in different types of hyperglycemic mice
The results above show that TMG-induced upregulation of O-GlcNAcylation significantly aggravated cerebral ischemia injury under hyperglycemic conditions. Next, we examined whether DON, a drug that downregulates O-GlcNAcylation, can antagonize the adverse effects of TMG on cerebral ischemic injury under hyperglycemic conditions. We injected 2.6μg DON via stereotaxic localization in the lateral ventricle 24 h before performing MCAO. Mortality, neurological function, and infarct size were measured to evaluate the degree of ischemic brain injury in the two types of hyperglycemic mice after DON treatment. Compared with the DM-sham group, mortality, behavioral scores, and infarct size were increased in DM-MCAO mice and decreased after DON injection (Fig. 6a, c, e). Similarly, mortality, behavioral scores, and infarct size in the HG-MCAO group were higher than that in the HG-sham group (Fig. 6b, d, f). Meanwhile, HG mice treated with DON had significantly reduced the size of cerebral infarcts, mortality and symptoms of neurological damage compared with that in the HG-MCAO group. These results suggest that inhibiting O-GlcNAcylation may diminish the HG-induced increase in BBB permeability.
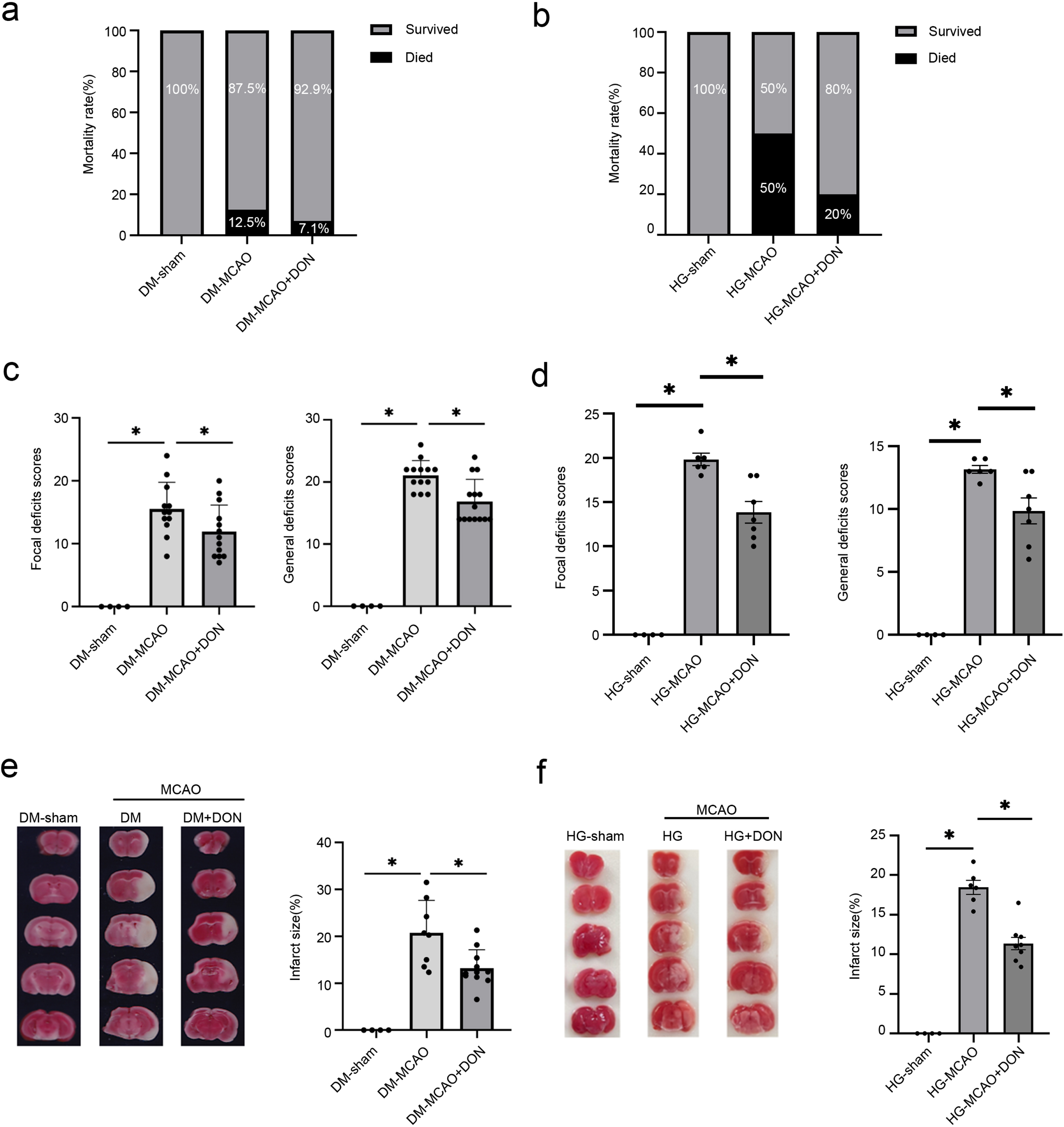
GFAT inhibitor DON alleviates cerebral infarction of cerebral ischemia in different types of hyperglycemic mice. (a-b) The graph depicts the death rate 24 h following MCAO. (c-d) Neurological scores in two types of hyperglycemia in the cerebral ischemia model. (e-f) Twenty-four hours after MCAO, coronal slices were stained with TTC, and the infarct volume was measured. Data are presented as mean±SD. *p < 0.05. DM-sham (n = 4), DM-MCAO (n = 12) and DM-MCAO+DON groups (n = 13); HG-sham (n = 4), HG-MCAO (n = 6) and HG-MCAO+DON groups (n = 7).
TMG promotes apoptosis in hyperglycemic mice subjected to ischemia-reperfusion injury
Over-upregulation of O-GlcNAc worsens ischemic brain damage under hyperglycemic conditions. Next, we explored the possible mechanisms through which protein O-GlcNAcylation exerts its effects. Our previous studies have confirmed that increased O-GlcNAcylation levels induce neuronal apoptosis [23]. Herein, we determined the expression of cleaved caspase 3, Bcl-2, and Bax, three apoptosis-representative proteins, to investigate the effect of O-GlcNAcylation on cell apoptosis in animal and cellular models of hyperglycemia with cerebral ischemia. In HG-treated bEnd3 cells subjected to OGD/R insult, an in vitro model of hyperglycemia with cerebral ischemia, the expression of pro-apoptotic proteins cleaved caspase 3 and Bax increased. In contrast, that of the anti-apoptotic protein Bcl-2 decreased. Notably, cleaved caspase 3 expression increased further, while the Bcl-2/Bax ratio decreased after TMG treatment (Fig. 7a, b). Compared to HG-treated cells, TUNEL staining demonstrated that the number of positive cells was increased after OGD/R insult, which was further increased in the HG and OGD/R-stimulated bEnd3 group after treatment with TMG (Fig. 7c, d). Similarly, in vivo, O-GlcNAcylation levels were remarkably increased in DM+MCAO+TMG mice compared to DM+MCAO mice (Fig. 7e, f). Significantly, compared with the DM-sham mice, the Bcl-2/Bax ratio decreased in DM-MCAO mice. The ratio was further considerably reduced after treatment with TMG, suggesting that TMG accelerates apoptosis in hyperglycemic ischemic mice (Fig. 7 g, h). These data indicate that the possible mechanism of O-GlcNAcylation in hyperglycemia-aggravated ischemic brain injury is related to promoting apoptosis.
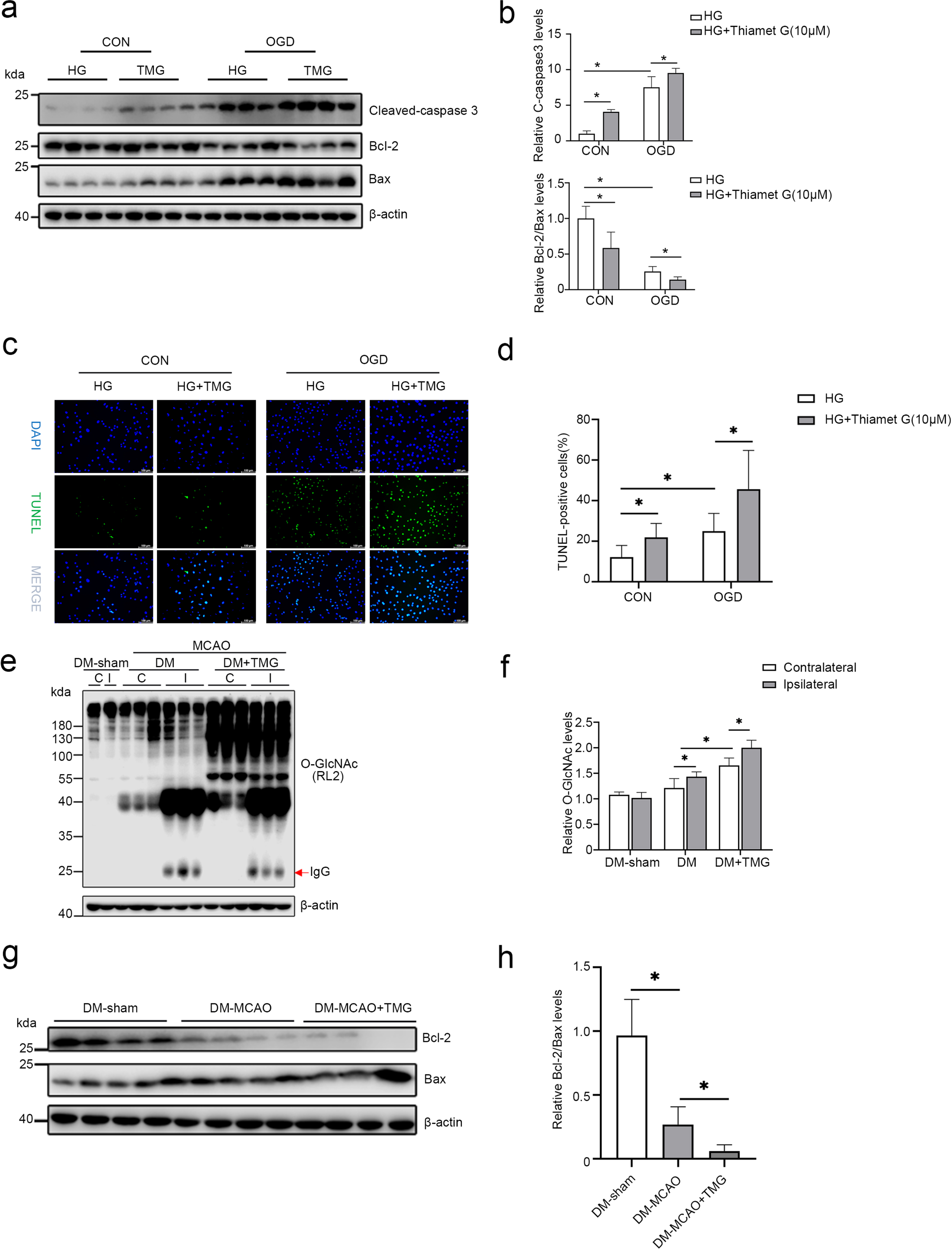
TMG promotes apoptosis in hyperglycemia with ischemia-reperfusion injury. (a-b) Western blot analysis of the levels of apoptosis-related proteins, cleaved-caspase 3, bcl-2, and bax in cells, followed by quantifying each band intensity and normalizing to the intensity levels of β-actin. (c-d) Representative immunofluorescence of TUNEL staining; green indicates apoptotic cells. Scale bar, 100μm. (e-f) Western blot analysis of protein O-GlcNAcylation using an RL2 antibody; quantification of all the blot bands is shown as the mean±SD in the graph. (g-h) Western blot analysis of Bcl-2 and Bax expression in DM-sham, DM-MCAO, DM-MCAO+TMG mice. β-actin was used as a loading control. All the quantification data are presented as the mean±SD (n = 3–4). *p < 0.05.
DISCUSSION
At least one-third of cases of AD exhibit different cerebrovascular disorders, micro- and macro-infarctions, and ischemic white matter changes are observed in patients with AD [28]. In the current investigation, in vitro and in vivo tests were performed for the first time to examine the impact of O-GlcNAc modification on cerebral ischemia damage under hyperglycemic conditions. Importantly, we propose a “double-edged sword” role of O-GlcNAcylation in cerebral ischemia, which suggests a novel, prospective therapy method for cerebral ischemic damage.
Compared with non-AD controls with similar risk characteristics, patients with AD may be at higher risk for stroke; what’s more, patients have a higher probability of hemorrhagic stroke [29]. Studies have shown that hyperglycemia-induced microvascular changes in the ischemic brain lead to increased permeability of BBB and even dysfunction [21]. However, vascular abnormalities are often ignored by researchers in AD.
After blood flow is restored, ischemic stroke patients with diabetes have more significant I/R damage, a worse prognosis, and a higher fatality rate than non-diabetic ischemic stroke patients [30]. Some researchers have discussed the impact of hyperglycemia on stroke in terms of post-stroke injury aggravation and induction of HT. After the stroke, patients have abnormal energy metabolism in the ischemic region of the brain, inflammatory cell infiltration, blood-brain barrier damage, and increased inflammatory response, resulting in oxygen radical release, vascular endothelial damage, and cell metabolic disorders, leading to the activation of apoptotic pathways and cell apoptosis. Hyperglycemia also aggravates vascular endothelial injury, promotes apoptosis, and BBB damage, inducing HT and seriously affecting the prognosis of stroke [31–35].
O-GlcNAcylation is an endogenous stress-activated response, and the critical factor determining the positive or negative effects of a specific O-GlcNAcylation modification is the type of injury. In acute or chronic injury, such as acute vascular injury or chronic hyperglycemia in diabetes, a modest increase in O-GlcNAcylation confers a survival advantage, whereas its excessive activation may be detrimental. A study of mice with diabetic cardiomyopathy suggests that an extreme rise in O-GlcNAcylation may interfere with other pro-survival signaling pathways, such as the Akt signaling pathway, desensitizing its cardioprotective effects [19, 37]. TMG, a specific OGA inhibitor, was utilized to upregulate protein O-GlcNAcylation to test its function in HG-induced cerebral ischemia damage. TMG is very stable when distributed in solution and can penetrate the BBB, making it appropriate for cell culture and animals [38]. It allows the manipulation of O-GlcNAcylation levels in neurological diseases. It has been shown that TMG dramatically decreased the extent of cerebral infarction, neurological abnormalities, and motor coordination deficiencies in ischemic animals [39].
We demonstrated that manipulating O-GlcNAcylation levels using drugs significantly affected cell survival and tissue damage in vitro and in vivo. Upregulation of O-GlcNAcylation levels in brain microvascular endothelial cells cultured in a medium containing normal glucose levels (5.56 mM) using TMG increased cellular resistance to OGD/R-induced damage. However, the culture of vascular endothelial cells for a long period under HG conditions resulted in chronic damage to the cells and a concentration-dependent decrease in cell viability. Furthermore, treatment of cells with TMG to upregulate O-GlcNAcylation levels resulted in concentration-dependent aggravation of OGD/R-induced hypoxic damage. This result is similar to those obtained in other studies [19]. In in vivo experiments, two animal models of hyperglycemia combined with stroke, a diabetic model combined with cerebral ischemia-reperfusion, and an acute hyperglycemia model following cerebral ischemia-reperfusion were used to explore the effect of O-GlcNAcylation. According to the experimental design, TMG, which crosses the BBB, was injected into mice intraperitoneally at a dose of 30 mg/kg 3 h before MCAO, according to a previous experimental study [18]. After the occurrence of cerebral ischemia-reperfusion, we focused on the damage in the group of hyperglycemic mice injected with TMG. The results showed a significant increase in O-GlcNAcylation levels in the brain tissue of normal and diabetic mice after cerebral ischemia and a further increase in O-GlcNAcylation levels in the brains of diabetic mice injected intraperitoneally with TMG (Fig. 7e, f). We found that both diabetic and hyperglycemic mice with ischemic injury had larger infarct areas, and TMG did not show neuroprotective effects in diabetic mice with cerebral I/R injury but further aggravated neurological deficits (Fig. 3h and Fig. 4d). Meanwhile, brain slices from diabetic mice or post-ischemic acute hyperglycemic mice showed speckled hemorrhage after stroke, and cerebral edema was evident. In contrast, brain slices from the TMG group showed high-density small dotted petechial hemorrhage, more pronounced cerebral edema, and a heavier occupancy effect (Fig. 3i and Fig. 4e). After TMG treatment, bands of 25 kDa appeared on the ischemic ipsilateral (infarct) side, suggesting that TMG may damage the blood-brain barrier and cause IgG leakage (Fig. 7e).
Hyperglycemia aggravates ischemic brain injury in two ways: on the one hand, it increases cerebral infarction size, and on the other hand, it induces HT. We used DON (an inhibitor of GFAT), which reduces HBP flow and O-GlcNAcylation, demonstrating the essential role of O-GlcNAcylation in hyperglycemia combined with stroke in mice [40]. DON (2.6μg), which cannot cross the BBB, was injected in the lateral ventricle 24 h before MCAO. We found that DON did not enhance ischemia-induced motor impairments or infarct size but significantly alleviated cerebral ischemic injury and HT in both diabetic mice and post-ischemic acute hyperglycemic mice (Fig. 5). It also mitigates mortality. However, we acknowledge that this study has some limitations. First, the beneficial effect on ischemia-reperfusion injury faded 48 h after drug injection, probably because of the small dose of the drug and the short duration of the drug’s life after injection; second, the lateral ventricular injection itself can cause damage to the brain, which is not in accordance with the clinical medical principles of treating patients by pharmacological intervention.
In a mouse model of ischemic stroke, O-GlcNAcylation facilitated apoptosis by inhibiting the phosphorylation/activation of AKT and the BCL2-associated cell death agonist (Bad). In this process, both Thr308 and Ser473 of AKT were modified by O-GlcNAc; however, overexpression of AKT in cultured cells attenuated O-GlcNAcylation-induced apoptosis. These findings imply cerebral ischemia induces O-GlcNAcylation, which induces apoptosis by downregulating AKT signaling [23]. Whereas in myocardial ischemia-reperfusion disease, also using agents that induce O-GlcNAcylation, O-GlcNAcylation decreased insulin-stimulated Akt phosphorylation and elevated the levels of apoptosis in cells, thereby attenuating the cardioprotective effects of insulin against I/R injury [19]. Indeed, our recent studies have revealed that increased O-GlcNAcylation in the cerebral ischemia model of normal mice was accompanied by increased apoptosis, which was further enhanced by a high dosage of TMG [23]. Therefore, we further investigated the mechanisms associated with ischemic injury in diabetic mice after the upregulation of O-GlcNAcylation levels from the perspective of apoptosis in both in vitro and in vivo experiments. The results demonstrated cleaved caspase 3 expression, and TUNEL-positive cells were further increased, whereas the Bcl-2/Bax ratio was decreased in TMG-treated cells. Similar results were obtained in vivo, where the levels of apoptosis were further increased, accompanied by an increase in O-GlcNAcylation levels after treatment of DM-MCAO mice with TMG (Fig. 7). It has been previously shown that a sharp rise in intracellular O-GlcNAc levels for a short period of time with different doses of TMG may lead to impaired mitochondrial oxygen consumption and complex metabolic activity [23]. It may be due to time- and dose-dependent changes in O-GlcNAcylation that affect mitochondrial function, triggering an endoplasmic reticulum stress response and ultimately driving the release of mitochondrial contents such as cytochrome c (Cyt-c) into the cytoplasm. HG significantly increases the release of endothelial AIF and Cyt-c into the cytoplasm, activating the P53 signaling pathway and triggering endothelial apoptosis [41–43].
Several studies suggest that increasing O-GlcNAcylation levels during acute ischemic injury can be protective. This conclusion has been demonstrated in ischemic diseases of the brain, heart, kidney, and small intestine [26, 44–46]. However, in our study of the effects of chronic hyperglycemic states on ischemic damage, we concluded, using in vivo and in vitro experiments, that upregulation of O-GlcNAcylation does not exert a protective effect in hyperglycemia-aggravated cerebral ischemia but rather exacerbates the damage. In contrast, decreasing O-GlcNAcylation levels ameliorated hyperglycemia combined with cerebral ischemic injury. Therefore, we propose a “double-edged sword” role for O-GlcNAcylation in cerebral ischemia. We further critically consider that the use of drugs may result in unforeseen off-target effects; consequently, genetic alteration of O-GlcNAc signaling is essential to circumvent this issue. We will continue to explore the role of protein O-GlcNAcylation in hyperglycemia-exacerbated cerebral ischemic injury and investigate the relevant mechanisms.
Studies have found that diabetes and ischemic stroke contribute to the development of AD [3]. In conclusion, our findings indicate that hyperglycemia-induced hyper O-GlcNAcylation may exacerbate brain I/R injury. It highlights O-GlcNAcylation as a viable therapeutic target for treating ischemic stroke patients to improve damage control and prognosis after cerebral ischemia, maintain the cerebrovascular system’s health in clinical stroke patients, and reduce the incidence of Alzheimer’s disease.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This study was supported by the National Natural Science Foundation of China, Nos. 81870941 (to JHG); the Nantong Commission of Health, No. MSZ2022048 (to HX) and QN2022045 (to RRS); Science and Technology Program of Nantong of China, No. MSZ2022097 (to JHG).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data presented in this study are available on request from the corresponding author.