
Abstract
Cerebral amyloid angiopathy (CAA) is characterized by amyloid-β aggregation in the media and adventitia of the leptomeningeal and cortical blood vessels. CAA is one of the strongest vascular contributors to Alzheimer’s disease (AD). It frequently co-occurs in AD patients, but the relationship between CAA and AD is incompletely understood. CAA may drive AD risk through damage to the neurovascular unit and accelerate parenchymal amyloid and tau deposition. Conversely, early AD may also drive CAA through cerebrovascular remodeling that impairs blood vessels from clearing amyloid-β. Sole reliance on autopsy examination to study CAA limits researchers’ ability to investigate CAA’s natural disease course and the effect of CAA on cognitive decline. Neuroimaging allows for in vivo assessment of brain function and structure and can be leveraged to investigate CAA staging and explore its associations with AD. In this review, we will discuss neuroimaging modalities that can be used to investigate markers associated with CAA that may impact AD vulnerability including hemorrhages and microbleeds, blood-brain barrier permeability disruption, reduced cerebral blood flow, amyloid and tau accumulation, white matter tract disruption, reduced cerebrovascular reactivity, and lowered brain glucose metabolism. We present possible areas for research inquiry to advance biomarker discovery and improve diagnostics.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a significant public health concern that warrants deeper exploration into its major contributors. The neuropathological features of AD include aggregation of extracellular amyloid plaques, intracellular hyperphosphorylated tau neurofibrillary tangles, and neurodegeneration [1]. Vascular dysfunction also is involved in all aspects of AD [2]; it promotes neurodegeneration via impaired cerebral perfusion [3], metabolic dysregulation [4], and inflammation [2, 5]. Vascular dysfunction can also change the structure and function of the vessels that comprise the blood-brain barrier (BBB) resulting in impaired glymphatic clearance of cytotoxic amyloid plaque and neurofibrillary tau and subsequent increased amyloid and tau accumulation in the brain [6, 7]. Cerebral amyloid angiopathy (CAA), a cerebrovascular condition in which amyloid-β aggregates in the cerebral vessel walls, is one of the strongest vascular contributors to cognitive decline and AD dementia [8]. In this review, we will discuss the relationship between CAA and AD, particularly, how neuroimaging markers can be used to study CAA alone and in the context of AD. We aim to improve understanding of the contributors to AD vulnerability for continued discovery of relevant AD biomarkers, effective therapeutics, and personalized medicine.
Relationship between CAA and AD
Although clinically distinct from AD, CAA is frequently observed in AD patients, appearing pre-clinically or comorbid with parenchymal amyloid and tau [9], and associated with advanced AD dementia symptomology and faster cognitive decline [10, 11]. However, CAA’s impact on AD risk, onset and progression are not well understood [8]. CAA may reduce the threshold for AD by inducing ischemia and reducing blood flow, impairing neuron survival and accelerating brain atrophy [12]. CAA may also potentiate parenchymal AD neuropathology through vascular injuries including ischemia, cerebral hemorrhages, and poor cerebral blood flow [13, 14]. These conditions contribute to disrupted white matter connections [15], accelerated cortical thinning [16], amyloid and tau deposition [17, 18], and eventual cognitive decline [13]. These relationships may also be bidirectional with early AD neuropathology driving CAA through vascular remodeling that damages glymphatic clearance and increases inflammation and oxidative stress to promote CAA development.
Etiology of CAA
CAA is a clinical condition in which amyloid accumulates in the small- and medium-sized arterial and sometimes capillary walls of the cortex and leptomeninges [19, 20]. Amyloid deposits progressively replace the medial layer of vascular smooth muscle cells and aggregate in the tunica adventitia (outer layer of the blood vessel wall), while simultaneously thickening the vessel wall [20, 21]. While CAA etiology is still unclear, CAA has been hypothesized to manifest from a convergence of disrupted glymphatic transport, failed interstitial fluid drainage and reduced arterial pulsations that stall the perivascular clearance of neuronal amyloid [12, 22].
CAA can be divided into two subtypes based on amyloid accumulation location. In CAA type 1 patients, amyloid is deposited primarily in the cortical capillaries with possible deposits in other blood vessel walls, while in CAA type 2 patients, amyloid is deposited primarily in leptomeningeal and cortical arteriolar and/or arterial walls with less likelihood of deposits in capillary walls [23].
CAA type 1 and 2 show differing relationships to AD. CAA type 1 is typically highly co-morbid with AD neuropathology. In addition to increasing AD risk, APOE4 increases the vulnerability to CAA Type 1 [24–26]. Like in AD patients, in CAA type 1 patients, Aβ42 is the predominant amyloid-β isoform that dysregulates neuronal signaling and aggregates to form amyloid plaques [27]. In contrast, those with CAA Type 2 are more likely to be carriers of the APOE2 allele [23, 28] which confers protection against AD. Although, both CAA types contain some mixture of Aβ40 and Aβ42 deposits [24, 29], CAA type 2 arterial deposition of amyloid predominantly consists of Aβ40. Aβ42 deposits are much less present in CAA type 2 [24, 29]. Further investigation that quantifies the number of Type 1 and Type 2 patients who subsequently develop AD is of importance.
Genetic and sporadic CAA
Genetic and sporadic forms of CAA are both characterized by amyloid deposition within small to medium sized arterial blood vessels and capillaries. Most CAA cases are sporadic with risk increasing based on age and low potency genetic variants [30]. Like AD, the APOE4 genetic variant is associated with an increased risk of CAA type 1 [24, 32], through structural and functional changes to the cerebral vasculature that impede amyloid-β clearance [32]. Other genetic variants in CLU, ABCA7, TREML2, and EPHA1 are also associated with the development of CAA- related neurovascular markers such as cortical superficial siderosis, lobar cerebral microbleeds, deep and periventricular white matter hyperintensities (WMHs), and centrum semiovale-enlarged perivascular spaces [33].
On the other hand, genetic or hereditary CAA emerges from single inherited genetic mutations that typically accelerate onset throughout early adulthood and mid-life [34–38]. Although hereditary CAA is rare, approximately six forms exist that are caused by various point mutations on the amyloid precursor protein gene, resulting in abnormal amyloid peptide formation and deposition [34]. Hereditary CAA includes the Dutch, Italian, Piedmont, Flemish, Iowa, Artic, and Icelandic type mutations [34]. Hereditary CAA types are heterogeneous in terms of severity of hemorrhages and parenchymal amyloid and tau accumulation. For instance, Dutch, Italian, Iowa, Piedmont, and Icelandic type patients experience frequent or recurrent hemorrhages and infarcts [34, 39–43], whereas Arctic type patients show low hemorrhage prevalence [43, 44]. Additionally, cognitive decline and early onset dementia manifest differently among hereditary CAA patients, arising from progressive neurovascular changes or repeat hemorrhagic strokes [34]. The Icelandic CAA patients show sparse parenchymal amyloid and tau [45] while Dutch, Italian, Iowa, Piedmont, and Flemish patients demonstrate a range of low to high amyloid plaque and neurofibrillary tau levels [34, 46]. Although the prevalence of hereditary CAA conversion to AD is understudied, certain hereditary forms of CAA are highly comorbid with early-onset AD and present with differing prevalence of neurovascular imaging markers. For example, Dutch and Italian types of CAA cause autosomal dominant early-onset AD comorbid with CAA [47, 48]. Frequent strokes and intracerebral hemorrhages in Dutch type CAA patients beginning in midlife are significant contributors to subsequent cognitive decline [49]. Iowa and Italian type mutations have been associated with a high distribution of cortical calcifications related to cognitive decline [50]. Flemish CAA patients form early-onset AD predominantly through Aβ40 deposits that aggregate into very large amyloid plaques [46]. Arctic CAA patients demonstrate brain atrophy and low parietal lobe blood flow associated with the presence of AD [44]. Further research is needed to evaluate the prevalence estimates of hereditary CAA conversion to dementia in those who have hereditary CAA. Down syndrome is also a genetic condition that leads to CAA onset. Down syndrome arises from an extra copy of chromosome 21 on which the amyloid precursor protein gene is located, and patients show CAA features associated with hemorrhages and strokes [51–53].
Clinical diagnosis of CAA
A definitive CAA diagnosis is possible only through postmortem autopsy [54–56]; however neurovascular imaging markers have allowed for “probable” and “possible” CAA diagnoses [57]. Autopsy studies have strengthened the connection between AD and CAA and have shown that increased CAA severity coincides with AD-related amyloid plaque accumulation [58]. However, reliance on autopsy for diagnosis limits the ability to diagnose in preclinical and early clinical disease states. Autopsy also does not allow the evaluation of disease staging and progression or examination of CAA’s relationship to cognitive decline [54]. Today, “probable” or “possible” CAA can be clinically diagnosed using the Boston Criteria version 2.0 (previous versions were entitled the Modified Boston Criteria and the Boston Criteria), a diagnostic rating system that combines evaluation of neuroimaging biomarkers and clinical examination data to specify the level of certainty for a CAA diagnosis [59]. The Boston criteria first introduced a set of criteria by which probable or possible CAA could be diagnosed using neuroimaging findings of cerebral hemorrhages and clinical information [55]. Later, the diagnostic criteria for CAA were updated to the Modified Boston Criteria to include the presence of blood products from cortical superficial siderosis to be counted towards the hemorrhage requirement for a diagnosis [57]. The Boston Criteria version 2.0 is the most recent diagnostic outline that improves the sensitivity and specificity of the CAA diagnostic rating scale to allow a broad continuum of cases to be included in the probable CAA diagnostic category [59]. In doing so, a probable CAA diagnosis can be made with a non-hemorrhagic marker that indicates vascular dysfunction. Current diagnostic criteria for probable CAA includes 1) the presence of multifocal cortical superficial siderosis without evidence of intracerebral hemorrhage or cerebral microbleed or 2) the presence of one white matter lesion with at least one lobar hemorrhagic marker [59]. Furthermore, the Boston criteria version 2.0 states that a possible CAA diagnosis can be made with one hemorrhagic lesion or one white matter feature, a change from the previous criteria that required a hemorrhagic marker. The inclusion of non-hemorrhagic markers in the probable and possible CAA diagnostic criteria allows for CAA patients in earlier disease stages who do not present with hemorrhages to be identified without compromising specificity [59]. Therefore, use of neuroimaging markers in the Boston Criteria version 2.0 can enable researchers to diagnose CAA across the spectrum severity and investigate its relationship with AD in living patients.
CAA and AD comorbidity prevalence
CAA is a common finding in AD patients. However, prevalence estimates of CAA vary based on study methodology and study sample [60]. A recent meta-analysis reported that estimates based on neuropathological exam suggest that 48% of AD patients have moderate to severe CAA, while MRI studies report that approximately 22% of AD patients have moderate to severe CAA. Prevalence estimates of CAA between neuroimaging and neuropathological studies are similar in cognitively normal older adults and intracerebral hemorrhage patients [60]. MRI studies may show lower prevalence rates given that up until the establishment of the Boston version 2.0 criteria, determination of CAA was based on the evidence of lobar microbleeds or hemorrhages resulting in an over-reporting of severe CAA that presents with lobar microbleeds and an under-reporting of mild and moderate CAA that emerges prior to microbleeds. Conversely, neuropathological studies may report higher rates given that post-mortem brain tissue analysis occurs in older adults who likely died from complications related to advanced CAA [60].
Bidirectionality of AD and CAA
Although this review focuses on the contribution of CAA to AD, the relationship between CAA and AD is likely bidirectional with early AD neuropathology potentially damaging vascular clearance mechanisms and predisposing individuals to develop CAA. AD neuropathology has been associated with cerebrovascular remodeling that alters the structure and function of the blood vessels. AD brains present with lower cerebrovascular density including reduced efficiency of tight junctions [61], greater basal membrane thickening [62] and vascular wall atrophy [63, 64]. Both amyloid-β and neurofibrillary tau exert toxic effects on cerebral blood vessels. For example, amyloid deposition has been associated with high levels of pro-inflammatory cytokines that induce neuro- and vascular inflammation and aberrant angiogenesis [63, 66]. Additionally, amyloid-β may increase oxidative stress and reduce antioxidant activity, making vascular endothelial cells more susceptible to apoptosis, inflammation, and related dysfunction [67]. Likewise, tau accumulation is associated with vascular dysfunction. For example, early Braak stage tau deposition in mild cognitively impaired (MCI) patients is associated with low cerebral blood flow and arterial stiffness that can potentiate arterial wall re-modelling and compromise glymphatic clearance [68, 69].
Gaps in the literature
Additional longitudinal prospective studies are necessary to understand the natural course of CAA. Predominantly, longitudinal studies of CAA have investigated the associations between cerebral microinfarcts, amyloid-β positivity [70], and future cognitive decline [70, 71]. For instance, CAA patients with cerebral microinfarcts who are amyloid-β positive have steeper memory decline across a three and a half year time span than CAA patients who are amyloid negative or present without cerebral microinfarcts [70]. These findings reflect previous results showing high microbleed count is associated with steeper decline in executive functioning, memory [71, 72], and processing speed [73] cognitive domains. Yet many long-term associations between neuroimaging markers and CAA progression have not been evaluated. There remains a need for longitudinal studies to investigate the onset and trajectory of neuroimaging markers related to BBB permeability, cerebral blood flow, cerebrovascular reactivity, white matter tract microstructure, and tau deposition and progression to evaluate their associations with cognitive decline and future dementia outcomes.
Non-CAA cerebral small vessel disease
Although CAA is the focus of this review, it is important to note that CAA is only one of many small vessel diseases that affects neurovascular injury and increases risk of AD. Other common non-CAA cerebral small vessel diseases include arteriosclerosis, hereditary small vessel disease, inflammation-mediated small vessel disease and venous collagen disease that incur damage to arterioles, capillaries and venules [74]. CAA and non-CAA small vessel disease patients also show overlapping neuroimaging markers [74] such as WMHs [75–78], intracerebral hemorrhages [79], and cerebral microbleeds [80, 81]. Non-CAA cerebral small vessel disease may increase AD vulnerability through vascular mechanisms. For example, non-CAA cerebral small vessel diseases damage the blood vessels and reduce blood perfusion to the neurons, depriving them of necessary oxygen and nutrients for survival, advancing apoptosis and reducing brain volume [82, 83]. Lacunar infarcts that commonly occur as a result of arteriosclerosis are associated with regional neuronal apoptosis [84] and AD-associated cerebrospinal fluid (CSF) levels of low tau and high Aβ42 [85, 86]. Non-CAA cerebral small vessel diseases such as Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, hereditary diffuse leukoencephalopathy, and venous collagen disease are also associated with high WMH burden [87] related to reduced cerebral blood flow, impaired cerebrovascular reactivity, disrupted BBB permeability and lowered white matter perfusion [85, 88–91].
NEUROIMAGING CHARACTERISTICS OF CAA
Although some of the most sensitive identifying neuroimaging characteristics of CAA are cortical superficial siderosis, lobar intracerebral hemorrhages, and cerebral microbleeds [92–94], CAA also promotes disease processes that contribute to neurodegeneration, AD neuropathology and cognitive decline [95]. Therefore, investigation of CAA using multimodal neuroimaging can help determine CAA staging and establish connections between CAA and AD. Given the implications of CAA in vascular injury, amyloid and tau aggregation, cerebral blood flow reductions, white matter structural decline, and hypometabolism, we will be discussing neuroimaging modalities commonly used to assess these features (Fig. 1).
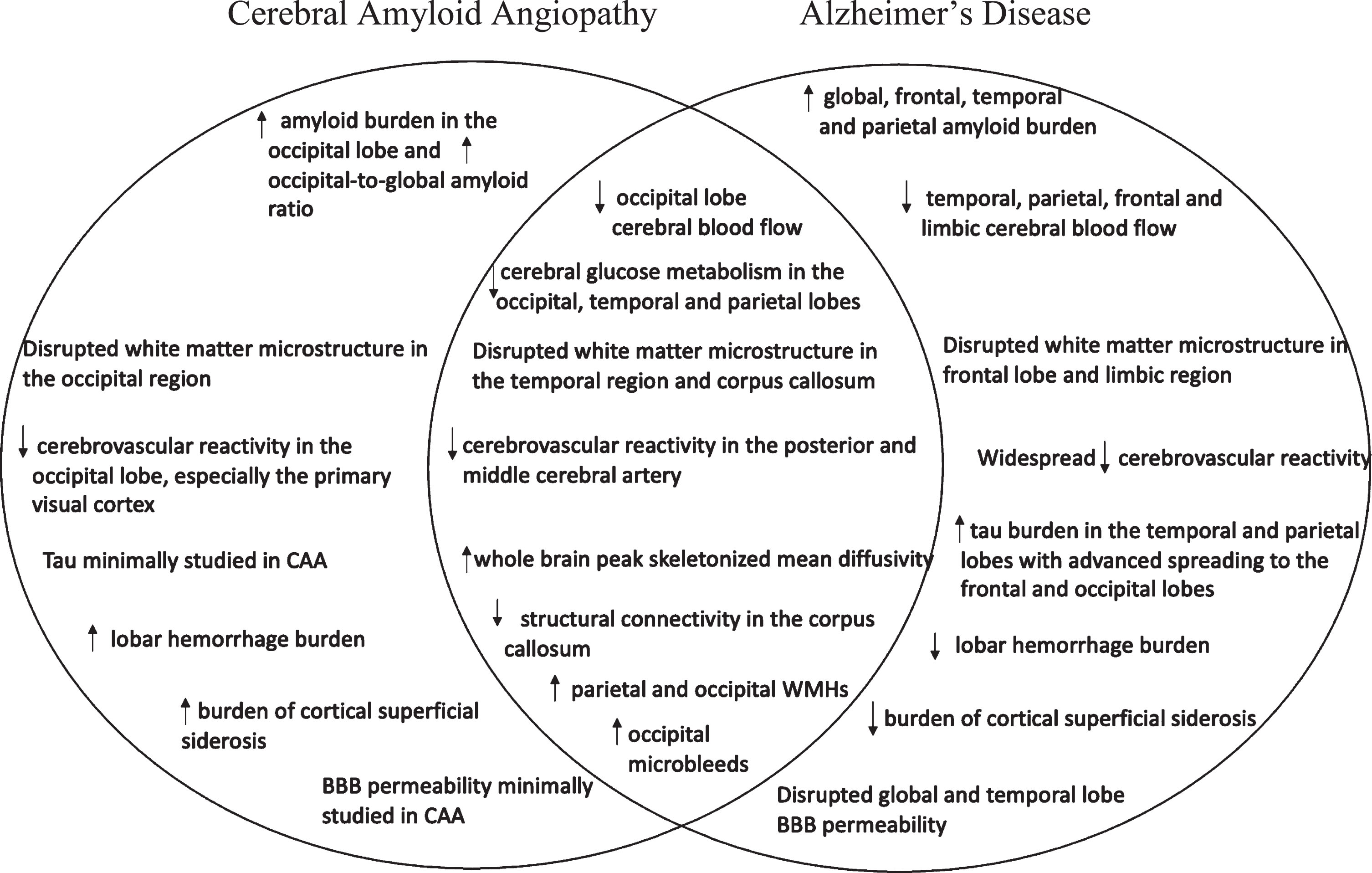
Overview of the shared and distinct neuroimaging markers of cerebral amyloid angiopathy and Alzheimer’s disease. CAA, cerebral amyloid angiopathy; BBB, blood-brain barrier; WMH, white matter hyperintensity.
T2* gradient echo recovery or susceptibility weighted images
As a result of the physical stress and neurotoxic effects imposed by amyloid on the blood vessels, CAA patients are at an increased risk for cortical superficial siderosis, cerebral microbleeds, and lobar hemorrhages [96, 97]. Susceptibility weighted imaging and T2* gradient recalled echo imaging are the best neuroimaging modalities to assess microbleeds, hemorrhages, and cortical superficial siderosis, with susceptibility weighted imaging being more sensitive for detecting small microbleeds than T2* gradient recalled echo [98–101]. Susceptibility weighted imaging and T2* gradient recalled echo identify paramagnetic properties of blood breakdown products that highlight the signal intensity differences between microbleed areas and surrounding tissues [102–105].
Cortical superficial siderosis
Cortical superficial siderosis is a pattern of blood-breakdown product, called hemosiderin, deposited on the convexities of the cortical sulci [106, 107]. Cortical superficial siderosis spares the brainstem, cerebellum, and spinal cord. Cortical superficial siderosis can be defined as either focal, in which a blood breakdown product is concentrated in a focused portion of the cortical sulci, or disseminated, in which a blood breakdown product is spread throughout cortical sulci [57, 107]. Specifically, the disseminated form, has a high co-occurrence with CAA and a lower co-occurrence in non-CAA or AD patients [57, 108]. Cortical superficial siderosis is a key neuroimaging marker of possible and probable CAA [109] and one of the strongest markers to determine a CAA-related hemorrhagic event and to predict future intracerebral hemorrhage risk [92, 110]. Greater cortical superficial siderosis severity has been associated with greater CAA severity in the leptomeningeal vessels [97]. Cortical superficial siderosis has also been associated with inflammation and may potentiate AD through inflammatory mechanisms. Histopathological study of possible and probable CAA brains found that macrophages surrounded regions of cortical superficial siderosis [97]. Additionally, upregulation of reactive astrocytes was proportionate to cortical superficial siderosis severity [97]. Moreover, CAA patients with high cortical superficial siderosis involvement showed AD-related spatial patterns of neurodegeneration. CAA patients with high cortical superficial siderosis burden showed lower hippocampal volume, and deficits in memory performance compared to CAA patients with low cortical superficial siderosis [111]. Additionally, CAA patients with high cortical superficial siderosis demonstrated greater AD-related parietal, temporal and frontal lobe atrophy in the precuneus, posterior cingulate, parietotemporal, superior frontal and medial temporal areas than healthy controls [111]. However, the role that cortical superficial siderosis plays in AD is unclear and likely very complex. Inoue and colleagues concluded that cortical superficial siderosis was scarce in AD patients [108]. However, it is unclear whether superficial siderosis mainly appears in AD patients as a result of concomitant severe CAA. For example, Jakel and colleague’s meta- analysis reported that the prevalence of cortical superficial siderosis appeared in approximately 5% of AD patients with moderate to severe CAA [60]. However, whether the distribution of cortical superficial siderosis more heavily fell in patients with severe CAA or was evenly distributed between patients with moderate and severe CAA was not reported. This leaves an unaddressed gap as to whether cortical superficial siderosis is observed in AD because of co-morbid severe CAA. Another meta-analysis found that although there is low prevalence of cortical superficial siderosis in patients with cognitive impairment, cortical superficial siderosis significantly predicts a clinical AD diagnosis [112]. However, it remains unclear whether the patients who were later clinically diagnosed with AD demonstrated neuropathological evidence of AD or instead had CAA alone accompanied by cognitive decline. It is suggested that although cortical superficial siderosis is not common in AD patients, it may affect AD progression, possibly due to the high correlation between cortical superficial siderosis presence and CAA-related cerebral microbleeds [108, 112]. Further investigation as to the independent or dependent associations between cortical superficial siderosis, CAA, and AD may help clarify the relationship between CAA and the advancement to AD [111].
Cerebral microbleeds
Cerebral microbleeds also commonly occur in CAA. CAA-related microbleeds tend to occur in small lobar arteries and arterioles of the posterior regions of the brain, especially the occipital lobe [23, 113] and posterior temporal lobes [22], whereas non-amyloid-related hemorrhages associated with hypertension or diabetes [114] tend to occur in the deep subcortical areas such as the thalamus, basal ganglia and infratentorial regions [22, 115]. This regional predilection of CAA microbleeds may help distinguish CAA from other small vessel diseases. CAA patients show a high number of cerebral microbleeds in the occipital and parietal lobes. Like CAA patients, cerebral microbleeds show a lobar topography in AD patients, especially in the occipital lobe [115, 116], appearing in high numbers in the interface between gray matter and white matter relative to healthy controls. Cerebral microbleeds increase the risk of ischemic stroke [117] but associations between microbleeds and cognitive decline have been non-significant and unclear [117–119]. In AD patients, the contribution of microbleeds to cognitive performance may be dependent upon several factors such as baseline cognitive impairment, medial temporal lobe atrophy and presence of other markers of vascular disease [10, 118]. The role of cerebral microbleeds has also been difficult to determine in CAA patients. However, a recent study indicated that CAA patients with cognitive decline show a higher prevalence of cerebral microbleeds especially in the temporal and insular lobes compared to CAA patients with hemorrhage-induced cognitive decline. This study suggests that cerebral microbleeds in specific lobar regions contribute to cognitive decline before the appearance of a hemorrhagic event [120]. Lastly, in addition to spatial distribution, number of cortical microbleeds may also distinguish AD patients both from comorbid AD and CAA patients and from healthy controls. De Reuck and colleagues demonstrated that co-morbid AD and CAA patients, have a greater number and wider distribution of cortical microbleeds and microinfarcts, particularly in the posterior brain regions such as the inferior parietal gyrus, precuneus, and cuneus, compared to AD and healthy control participants [121].
Lobar hemorrhages
Lobar hemorrhages commonly occur in the cortical regions of patients with advanced CAA [122]. Although, the spatial distribution of CAA hemorrhages in the temporal and occipital lobes have been suggested as defining characteristics of CAA [123], recent findings have demonstrated that lobar intracerebral hemorrhage is not a specific marker for CAA given its prevalence in other cerebral small vessel diseases like arteriosclerosis and mixed cerebral small vessel disease. For example, only 10% of lobar intracerebral hemorrhage patients have pure CAA while 50% have mixed CAA and hypertensive arteriopathy [124]. The remaining 40% have only hypertensive arteriopathy [124]. Additionally, CAA-related microbleeds in older adults may be comorbid with microbleeds from another etiology such as traumatic brain injury [105]. Lastly, patients with CAA with no lobar hemorrhages remain challenging to diagnose. Although updated, the recent Boston 2.0 criteria only has 50% diagnostic sensitivity for CAA patients without intracerebral hemorrhage [59].
Considering that vascular dysfunction is a pathological hallmark of CAA and AD, it is unclear why intracerebral hemorrhage prevalence is higher in CAA patients than AD patients. Potentially, these differences in intracerebral hemorrhage frequency stem from differences in vascular versus parenchymal amyloid at the BBB. For example, based on immunohistochemical analysis of CAA brain tissue, Jakel and colleagues found that vascular amyloid colocalizes with an upregulated expression of matrix metalloproteinase-9 (MMP9), a BBB-related protein associated with the degradation of the extracellular matrix and implicated in BBB disruption and intracerebral hemorrhage [125]. On the other hand, parenchymal amyloid does not show associations with upregulated expression of MMP9. Levels of MMP9 are especially high in CAA brains with intracerebral hemorrhages [125]. Potentially, AD patients present with a different MMP9 expression signature given the higher abundance of parenchymal amyloid compared to vascular amyloid, resulting in lower intracerebral hemorrhages relative to CAA patients [125]. Further research into mechanisms that underlie the differences in BBB permeability and hemorrhage frequency between CAA and AD should be performed.
Blood-brain barrier permeability and dynamic contrast enhanced MRI
Dynamic contrast enhanced MRI (DCE-MRI) measures the permeability of the BBB, a biomolecular semipermeable network of cells that prevents neurotoxin influx and facilitates waste efflux [126, 127] in living beings. Disruption of the BBB through amyloid-related weakening of the cerebral vessel walls is a significant consequence of CAA [128] and may be an initial pathological event in AD [129]. Cerebral microbleeds related to CAA may also lower the amyloid-β transporter expression in the BBB, resulting in lower amyloid clearance and higher parenchymal aggregation [105, 130]. For example, autopsies of CAA brains have shown significantly greater BBB disruption in amyloid burdened vessels than vessels free of amyloid [131]. DCE-MRI quantitatively assesses the rate of contrast agent seeping from the cerebral blood vessels into the brain’s extravascular spaces [132]. DCE-MRI may be sensitive to the breaches of BBB permeability [132], a potential contributor to cognitive decline and AD. For instance, in patients with vascular mild cognitive impairment, higher BBB leakage rate in the white matter and volume of WMHs are both correlated with global cognitive deficits [133]. Similarly, MCI patients and APOE4 carriers at risk of AD show heightened permeability of the hippocampus and medial temporal lobe [134–136] with AD patients demonstrating greater whole brain BBB permeability disruptions compared to healthy controls [137, 138]. Investigations of BBB permeability and cognitive performance is an understudied area in CAA research. Limited studies have used DCE-MRI to investigate BBB permeability in probable CAA patients. Aksoy and colleagues studied patients who had experienced spontaneous hemorrhages which included a subsample of five patients presumed to have probable CAA. Probable CAA patients presented with more permeable BBB compared to patients with hemorrhages from a presumed chronic hypertensive etiology [139]; additionally, all patients with hematomas demonstrated higher BBB permeability in regions proximal to their hematoma site [139]. This work was the first to investigate an important topic but given the small sample size of only five probable CAA patients, investigations should be replicated in a larger sample before its results can be confidently applied to the greater CAA population. It is also worth noting that patients investigated in this study experienced an acute hemorrhage and not chronic hemorrhages that may be more strongly associated with BBB breakdown. Given that Dutch type CAA patients experience recurrent hemorrhages, investigation with Dutch type CAA patients may be a better demographic to investigate BBB permeability associations between CAA as well as longitudinal associations between BBB permeability changes and AD progression and cognitive decline in CAA patients. Study of the permeability of the BBB in CAA patients prior to hemorrhage also warrants further investigation. Future research evaluating the associations between CAA, BBB permeability, and AD onset is needed.
Cerebrospinal fluid flow and DCE-MRI
Along with other neuroimaging measures such as phase contrast MRI [140], DCE-MRI may also be used to investigate CSF flow dynamics. This area may be relevant in understanding CAA etiology as impaired glymphatic transport systems may impede appropriate CSF flow [141]. Chen and colleagues used DCE-MRI to demonstrate that lower CSF drainage occurs along the carotid arteries in CAA transgenic animals, potentially as a result of lower functionality of the upstream glymphatic pathway [142]. As it currently stands, DCE-MRI has not been used to identify or differentiate CAA patients from AD patients or control participants. However, DCE-MRI may be a useful tool to investigate and identify the brain regions vulnerable to altered fluid dynamics that affect CAA onset.
Amyloid deposition and amyloid PET
Amyloid positron emission tomography (PET) can be used to quantify and localize amyloid deposition related to CAA and AD. CAA is associated with high levels of vascular amyloid deposition due to damaged perivascular clearance [143]. Amyloid PET uses a radioligand to tag amyloid plaques in both cerebral vessels and the cortex [144]. Although, amyloid PET imaging cannot differentiate between radiotracer binding in the vascular and cortical regions, amyloid PET signal of probable CAA patients is significantly greater than that of healthy controls and patients with other small vessel diseases [17, 145–147], and is greatest in regions proximal to hemorrhages of probable CAA patients [145]. Amyloid PET studies have provided insight into spatial characteristics of amyloid retention in probable CAA patients [148] and have underscored similarities and differences between probable CAA and AD patients. Most consistently, CAA patients demonstrate high amyloid load in occipital regions [17]. For example, in comparison studies between probable CAA and AD patients, CAA patients exhibit higher amyloid burden in occipital regions and higher occipital-to-global amyloid ratio than AD patients [146, 150]. Conversely, AD patients demonstrate higher global [146, 149] and frontal-global uptake ratio than CAA patients [146] as well as higher temporal and parietal amyloid retention compared to healthy control participants [149, 151]. Planton and colleagues also compared amyloid retention between CAA and MCI patients and found that MCI patients show higher amyloid uptake in parietal and temporal regions compared to CAA patients [151]. These findings are consistent with other studies that show that high regional amyloid retention in temporal and parietal cortices is predictive of MCI conversion to AD [152]. Autopsy findings have similarly found the parietal cortex to be a high concentration area of vascular amyloid for patients with comorbid CAA and AD in comparison to patients with either “pure” CAA or AD [153]. Amyloid PET may be especially useful to chronicle staging of amyloid progression in CAA patients [154].
Tau deposition in CAA and tau PET
Tau PET imaging is a neuroimaging modality that uses a radioligand to label pathological tau [155]. Animal models [156] and post-mortem human studies of CAA have shown tau accumulation near CAA-predominant regions [157, 158]. The relationship between CAA and tau is still unclear but may be bidirectional in which vascular amyloid may either trigger tau or tau may trigger vascular amyloid. For instance, blood vessels burdened with vascular amyloid may enhance glial and inflammatory cytokine activation [159] that jumpstarts tau aggregation [8]. Alternatively, tau deposition from neurons may initiate arterial remodeling that hampers amyloid clearance resulting in CAA-related amyloid accumulation [68]. Tau is a hallmark protein of AD that is closely correlated with neuronal dysfunction and poor cognitive performance [160]. Tau typically follows a topographical pattern of spread in which tau begins aggregation in the medial temporal lobe and spreads to neighboring temporal, parietal, occipital and frontal cortical regions as AD worsens [161, 162]. Advancing tau deposition across the brain is associated with AD-related grey matter atrophy, poorer cognition [163] and greater global amyloid burden [164]. Tau PET can distinguish between MCI, AD, and healthy control participants based on tau progression across the Braak stages. Braak staging is a system that characterizes the progression of tau spreading throughout the cortex along the AD trajectory [165–167]. For instance, AD patients show higher tau uptake in the Braak stage V and VI regions compared to healthy controls [168]. Although tau PET studies have been limited in investigating CAA, Schoemaker and colleagues demonstrated that tau deposition in cognitively impaired probable CAA patients may resemble an AD-like pattern. Additionally, this AD-like tau staging in CAA patients is also associated with worse cognitive performance. Probable CAA patients with memory impairments exhibited greater tau PET signal in AD signature temporal and parietal regions, including the entorhinal cortex, fusiform gyrus, temporal gyri, parietal lobules, posterior cingulate, and precuneus compared to probable CAA patients without memory impairments [18]. No published studies have compared tau PET signal between CAA and AD patients. A study of this nature would be a valuable next step in understanding the relationship between tau and CAA and tau’s underlying mechanistic role in the trajectory from CAA to AD.
CSF biomarkers of total tau and tau phosphorylated at threonine 181 (ptau181) may also be used to distinguish CAA from AD [169]. Elevated concentrations of soluble total tau and ptau181 are both found in AD patients [169]. Total tau is a non-specific general indicator of neurodegeneration [170] and ptau181 is a specific biomarker of AD related changes [171]. Analyses of CAA, AD patients, and healthy control participants showed that CSF total tau and ptau181 were both significantly higher in AD patients than in probable CAA patients and controls while only CSF total tau in probable CAA patients was significantly greater than in controls [169, 172]. Given that CSF changes of tau occur before PET changes, studies combining CSF tau biomarkers and tau PET would help clarify tau staging in CAA patients and the effect of tau on cognitive decline and AD development.
Autopsy studies have demonstrated high levels of parenchymal tau deposits in CAA patients. Brain samples from CAA patients demonstrated higher Braak staging and lower pre-mortem cognitive scores relative to brain samples from people without CAA [173]. A recent study that analyzed autopsied patient data from the Rush Memory and Aging Project, the Religious Orders Study, and the Minority Aging Research Study showed that among severe CAA patients, higher parenchymal amyloid plaque levels were positively associated with higher tau levels [174] and that tau mediates the association between CAA and cognitive impairment in CAA patients with high amyloid levels [174]. However, there are challenges in relating cognitive impairment to autopsy measures as the last cognitive assessment acquired may or may not be close to time of death. Given that tau PET has been a largely unexplored neuropathological marker in CAA, further analyses that relate CAA and tau deposition to longitudinal cognitive performance and evaluate interactions between tau and CAA on AD development would be informative.
White matter hyperintensities in CAA and T2 FLAIR
T2-weighted fluid attenuated inversion recovery is one of the most sensitive neuroimaging modalities to measure markers of subacute microvascular ischemia and hypoxia known as WMHs [175, 176]. WMHs are a common neuroimaging finding that occur in healthy aging individuals and patients with vascular risk. WMHs are non-specific indicators of vascular dysfunction and appear in patients with both CAA and non-CAA cerebral small vessel diseases [177]. CAA compromises vasomotor function leading to lower blood flow to the white matter tracts and promotes vascular lesions such as WMHs [23]. T2- weighted fluid attenuated inversion recovery suppresses CSF signal to increase the contrast between WMHs and the surrounding white matter tissues [178, 179]. T2- weighted fluid attenuated inversion recovery imaging studies of WMH associations with CAA revealed that WMH volume increases with CAA progression and cognitive impairment [180], a finding that has been demonstrated in MCI patients in which high WMH volume accelerates risk for cognitive decline and dementia conversion [181]. Similar to CAA patients, AD patients demonstrate larger WMH volumes that appear in periventricular and posterior regions [78, 182–184], including the parietal and occipital lobes [183, 186]. Conversely, WMH volumes appear larger in probable CAA patients than in healthy controls [184]. Although, WMH spatial distribution and volume are similar in CAA and AD, WMH spatial distribution patterns may help differentiate CAA from other small vessel diseases. For example, although both CAA and hypertensive arteriopathy are associated with high total WMH count, WMHs are typically found in subcortical white matter regions in probable CAA patients whereas hypertensive arteriopathy patients show WMHs in the peri-basal ganglia [77].
Structural connectivity of white matter tracts and diffusion tensor imaging
Diffusion tensor imaging is used to interrogate white matter microstructure organization and disruption by evaluating the rate of diffusion of water molecules [187]. CAA is related to obstructed blood flow to the white matter tracts, which can bring about microstructural changes that disturb the short- and long-range connections of different circuitries and affect efficiency of the brain’s networks [15, 188]. Transgenic animal models of CAA have revealed that white matter tracts proximal to occluded blood vessels experience myelin loss [189], contributing to disturbed white matter structure and communication between brain regions. Metrics such as mean diffusivity (MD) and fractional anisotropy (FA) are common sensitive indicators to distinguish changes in axonal degeneration and myelin loss [190]. Interrogation of white matter tract changes may be beneficial in distinguishing diseases. For instance, FA values of cerebral microbleed patients from a mild traumatic brain injury were significantly higher than FA values of cerebral microbleed patients from a non-traumatic brain injury etiology [191]. Autopsy examinations have corroborated the regional white matter tract disruptions in CAA by showing low FA and high MD being correlated with characteristics of microstructural degeneration such as less dense tissue, lower myelin density, and lower axonal density [190]. Most consistently, probable CAA patients show white matter tract changes predominantly in the occipital, parietal and posterior temporal tracts of the brain [15, 192]. Poor structural connectivity in the posterior regions of the brain is associated with occipital lobe thinning [193], lower brain volume, higher vascular markers, greater amyloid load, and poorer cognitive performance compared to healthy control participants [192]. Structural connectivity changes in the frontal tracts and between the posterior and frontal tracts occur as CAA severity worsens [188, 190]. Similar to CAA patients, AD patients also show reduced connectivity in the corpus callosum [15, 195], a major white matter tract that connects the brain hemispheres. Comparison between AD and CAA patients demonstrate that AD patients show higher diffusivity in the fornix, the output tract of the hippocampus, than CAA patients [196], possibly due to the significant hippocampal damage that occurs during AD. Additionally, AD patients demonstrate disrupted association pathway connections such as the inferior and superior longitudinal fasciculus and uncinate fasciculus, and lower FA values in the cingulum, parahippocampal, cerebellum, and thalamic white matter tracts [195, 197–205] compared to control participants. Therefore, interrogation of white matter structure can lend insight into common widespread and local neuronal communication failures and neurodegenerative processes correlated with CAA, AD, and cognitive impairment.
Peak width of skeletonized mean diffusivity (PSMD), a measure derived from diffusion-weighted MRI, can also be used to assess white matter tract microstructure disruption across the whole brain. PSMD is calculated by creating a skeletonized version of the MD values across the main white matter tracts [206]. Higher values are generally associated with less restrained water diffusion, which is common in many diseases and disorders [206]. PSMD has shown several benefits in contrast to the use of voxelwise MD and FA values. Particularly, PSMD is calculated automatically, avoids the influences of partial volume effects that arise in atrophied brains, and does not require spatial smoothing, which can add variability to results [207]. PSMD values have consistently been higher in probable CAA patients than healthy control participants [208–211]. Patients with comorbid CAA and MCI also show increased PSMD compared to MCI patients without CAA [212], suggesting that CAA may be associated with widespread white matter microstructure effects. However, in comparison to AD patients, McCreary and colleagues showed that PSMD values were similar to values found in CAA patients [209].
Cerebral blood flow using arterial spin labeling
Arterial spin labeling measures tissue perfusion throughout the brain and is the most promising neuroimaging tool for mapping blood flow [213]. CAA and CAA-related hemorrhages can contribute to capillary occlusions and impair vasodilation to impede blood flow within the brain [214, 215]. Arterial spin labeling magnetically labels water within the arterial blood to trace the rate of blood delivery to the cerebral capillaries [216, 217]. Arterial spin labeling provides an advantage over other neuroimaging measures, such as functional MRI, in that it approximates the location and intensity of neural activity to provide absolute quantitative measures of resting cerebral blood flow rather than a relative measure [218]. Lower regional cerebral blood flow may be a marker of CAA severity and impending cognitive decline. Cerebral blood flow measurements in symptomatic Dutch type CAA patients demonstrated lower blood flow in the occipital lobe and basilar artery compared to healthy controls and pre-symptomatic Dutch type CAA patients [219]. However, nondemented CAA patients demonstrated similar cerebral resting blood flow measurements compared to controls [220] suggesting that cerebral blood flow changes may not appear until considerable damage or early cognitive symptoms emerge. Relative to control participants, MCI and AD patients demonstrate regional decreases in cerebral blood flow in extensive brain regions, including the parietal, temporal, frontal, occipital, and limbic regions [221–225]. However, AD studies have shown mixed results with some findings indicating that cerebral blood flow may increase in the frontal lobe and limbic structures of MCI patients before AD onset [226]. Additional studies are needed to incorporate longitudinal measurements of cerebral blood flow patterns to uncover potential staging of cerebral blood flow in CAA patients over time to explore the relationships between cerebral blood flow changes related to CAA and cognitive decline.
Although imaging studies of cerebral blood flow have been limited, autopsy studies have investigated microinfarcts and ischemia as a result of CAA-related small vessel occlusions and cerebral blood flow reductions [227]. Capillary type CAA and severe CAA were independently associated with infarcts in vulnerable brain regions such as the CA1 subregion of the hippocampus [227]. More research is needed to investigate the effects of cerebral blood flow changes on AD-relevant neuroimaging markers in CAA patients.
Cerebrovascular reactivity
CAA inhibits the ability of cerebral arterioles and arteries to dilate and deliver blood appropriately to metabolically active neural substrates, a process known as cerebrovascular reactivity [228]. Cerebrovascular reactivity is characterized as a vasodilatory response to the change in extracellular pH levels [229]. Cerebrovascular reactivity involves measuring cerebral blood flow responses after a hypercapnic challenge such as a breath holding or carbon dioxide inhalation test [229]. Methodologies that measure cerebrovascular reactivity include transcranial doppler ultrasound and blood oxygen level-dependent (BOLD) functional MRI (fMRI). Transcranial doppler ultrasound measures cerebral artery blood velocity using cerebral circulation and pulsatility index [230, 231]. Functional MRI also assesses cerebrovascular reactivity patterns by measuring BOLD activity while the participants are exposed intermittently to different carbon dioxide levels either externally or through guided breath holding [232, 233]. Signal from BOLD fMRI in humans arises largely from veins and capillaries rather than from arteries [234]. Lower cerebrovascular reactivity in probable CAA patients has been proposed as a biomarker for early detection of CAA [219, 220].
Transcranial doppler and CAA
In response to a breath-holding task, studies using transcranial doppler have shown that both probable CAA and AD patients have lower reactivity in the posterior cerebral artery (PCA) and middle cerebral artery compared to healthy controls [235]. Similarly, in comparison to controls, probable CAA patients demonstrated lower blood flow velocity in the PCA in response to a visual task but no difference in the middle cerebral artery in response to a carbon dioxide challenge [236]. Reinhard and colleagues also showed lower cerebrovascular reactivity in the PCA of “pure CAA” patients compared to controls, which was correlated with number and severity of microbleeds, while middle cerebral artery cerebrovascular reactivity was comparable between CAA patients and healthy controls [237]. Further investigation of cerebrovascular reactivity in probable CAA patients is warranted given the small sample sizes of previous studies [236, 238] and limited comparison analyses between CAA and AD patients. It is possible that because vasculature within the occipital lobe is a preferential region for amyloid deposition and subsequent microbleeds, that the PCA that primarily supplies blood to the occipital lobe, is a primary affected region.
Functional MRI and CAA
Hypercapnia induces vasodilation through the increase of carbon dioxide in the blood [229]. CAA and AD patients both show lower global cerebrovascular reactivity; however, region-specific depression in cerebrovascular reactivity has been observed in the visual cortex of CAA patients [238]. In comparison to healthy controls, CAA patients show significantly smaller activation regions and lower BOLD amplitude in the posterior and middle cerebral arteries in response to a hypercapnic challenge [238]. Beaudin and colleagues compared whole brain BOLD fMRI responses to hypercapnia between CAA, AD, MCI patients, and healthy controls and found that CAA and AD patients show lower cerebrovascular reactivity in gray matter, white matter, global and AD-related regions such as the posterior cingulate, precuneus, middle temporal gyrus, and lateral occipital cortex compared to healthy controls [228]. In particular, CAA patients showed the lowest cerebrovascular reactivity in the primary visual cortex suggesting the visual cortex is a particularly vulnerable region for CAA-related cerebrovascular reactivity deficits [228]. While CAA patients have consistently shown compromised cerebrovascular reactivity specifically in the occipital lobe and posterior brain regions, most studies have shown that AD patients have a lower cerebrovascular reactivity response in more widespread regions, including the occipital lobe, compared to controls [239–243].
Resting state-fMRI
Resting state-fMRI has been used to assess functional connectivity between neural networks in probable CAA patients. Resting state-fMRI evaluates functional networks that are synchronously active in varying brain regions throughout a scan while a participant is not engaged in an effortful task [244, 245]. Drenth and colleagues found that compared to controls, Dutch type mutation CAA carriers showed less functional connectivity within and between brain regions that comprise the default mode, visual, executive control, and frontoparietal networks [246]. Furthermore, symptomatic mutation carriers had even lower connectivity compared to the total CAA patient sample. Disrupted functional connectivity is a common finding in AD and MCI patients as well. Most consistently, AD patients show decreased resting state functional connectivity in the posterior cingulate cortex within the default mode network compared to healthy controls [247–250].
Visual task-based fMRI
Task-based fMRI evaluates how brain activity changes while a task or activity is being performed by the participant versus when the participant is at rest. Considering that deficits in cerebrovascular reactivity are pronounced in the occipital lobe of CAA patients, visual tasks are commonly used in fMRI studies to estimate BOLD responses evoked by a visual task in probable CAA patients [219, 251]. Probable CAA patients have shown significantly lower visual BOLD response amplitudes after presentation of a visual task compared to AD and MCI patients and healthy controls [251]. Both cross-sectional and longitudinal studies have shown, after visual stimulation, probable CAA patients displayed a smaller and more delayed hemodynamic response relative to healthy controls, indicating greater impairment of activation and regulation that significantly worsens over time [219, 238].
FDG-PET
Fluorodeoxyglucose-PET (FDG-PET) evaluates resting state cerebral glucose uptake that partly reflects synaptic activity [252]. CAA and CAA-related microbleeds can impact astrocytic glucose transport and neuronal glucose uptake that affects brain metabolism [253, 254]. FDG-PET uses a radiotracer with a similar molecular structure to glucose except active neurons do not metabolize the tracer [255, 256]. Cerebral glucose uptake rate is estimated by regional tracer retention [255].
A few studies have observed significant brain hypometabolism in probable CAA patients compared to healthy controls and AD patients. Bergeret and colleagues found that FDG-PET signal was lower in posterior parietal, temporal, occipital, and cingulate regions in probable CAA patients compared to controls [253]; whereas compared to AD patients, glucose metabolism in the occipital/posterior cingulate was significantly lower among probable CAA patients [257].
Microbleeds and hemorrhages likely impair glucose uptake and diminish FDG-PET signal by reducing BBB expression of glucose transporters that transport glucose and FDG to the brain [258]. Widespread ischemia and hypometabolism can exacerbate brain atrophy in probable CAA patients [259]. Samuraki and colleagues found that compared to AD patients with microbleeds unrelated to CAA, AD patients with CAA-related microbleeds demonstrated lower glucose metabolism and atrophy in AD-related regions such as the temporal and frontal lobes [259]. These results suggest that CAA-related microbleeds in the frontal and temporal lobe regions may contribute to AD severity and progression. AD patients have consistently shown lower cerebral glucose metabolism in the posterior parietal, temporal [260–262], and occipital lobes compared to healthy controls [261, 263] with some reports of increased cerebral glucose metabolism in the frontal lobe of AD patients [261]. Although there may be some regional variability in cerebral glucose metabolism depending on the stage of cognitive decline, long term hypometabolism throughout the brain is suggested to be a relevant predictor of impending dementia onset [264]. Studies investigating CAA using FDG-PET are sparse and contain small sample sizes; further investigation into CAA-related glucose metabolism disturbances with larger samples would be ideal.
CONCLUSIONS
Neuroimaging of CAA is an important step in understanding the underlying vascular mechanisms that contribute to neurodegeneration, AD neuropathology and cognitive decline. As more studies focus on using multimodal imaging, a clearer picture may illustrate how long-term vascular issues influence brain changes and dementia development. Longitudinal studies are needed to advance the field’s understanding of the series of events that lead to CAA and drive the connection between CAA and AD. Greater sample sizes may also equip researchers with the statistical power to investigate varying relationships between vascular and neurodegenerative mechanisms and cognitive outcomes. Additionally, studies that validate neuroimaging biomarkers of probable CAA in patients who later have autopsy-verified CAA could be conducted.
Advanced neuroimaging technology that increases spatial resolution can also provide information about vascular processes that affect neural dysfunction. High spatial resolution MRI allows for better visualization of the structure and function of affected perforating cerebral arteries and small vessels to improve sensitivity and specificity of CAA detection and diagnosis [265–267]. For example, 7Tesla ultrahigh field MRI provides better detection of lobar microbleeds in patients with CAA than 1.5Tesla MRI scans [268]. Furthermore, higher resolution of 7Tesla scans promotes detection of vascular iron accumulation and calcification in the occipital cortical regions of probable CAA patients, a biomarker of CAA verified by histopathologic examinations [269].
Deep learning tools can improve the ability to detect microbleeds and differentiate them from other vascular conditions through reduction of false positives and enhanced detection sensitivity [270]. For instance, research groups have started the development of customized deep learning tools that employ convolutional neural networks for automated detection and classification of hemorrhages [271]. These tools have been proposed to assist clinicians with CT-based diagnosis and differentiation of hypertensive-related hemorrhage versus CAA-related hemorrhage [271]. However, additional testing is needed to establish the accuracy and reliability of such automated methods.
Machine learning models also can use aggregated information to make useful predictions about CAA trajectory [272]. In patients with neurovascular MRI markers of CAA, amyloid PET positivity demonstrates worse prognosis than amyloid PET negativity [272, 273]. Gradient boosting machine and random forest algorithms identified imaging markers and clinical characteristics that are most predictive of amyloid PET positivity in probable CAA patients [272]. Both models determined clinical cutoff values for the most significant predictive variables of amyloid PET positivity including age, number of lobar cerebral microbleeds, deep cerebral microbleeds, lacunes, and microbleeds in the dentate nucleus [272]. Integration and implementation of these predictive models into clinical settings may assist clinicians who only have access to demographic information and neurovascular MRI markers to determine prognoses for patients with probable CAA.
Much effort has been devoted to developing immunotherapies to rid the brain vasculature of amyloid and restore vascular function. To assess efficacy of new therapeutic treatments, clinical trials have implemented functional neuroimaging markers as primary end points, specifically cerebrovascular reactivity. Transgenic CAA mouse models that have tested the efficacy of the experimental amyloid-β clearing therapeutic, Taxifolin, found that cerebrovascular reactivity was impaired in untreated CAA mice but rescued in CAA mice treated with Taxifolin [274, 275]. Similarly, human subjects’ clinical trials used cerebrovascular reactivity via fMRI BOLD activity to test the efficacy of the first CAA anti-amyloid immunotherapy, Ponezumab, as the primary endpoint in a cohort of probable CAA patients; however, no significant differences were found [276]. Potentially, further exploration of neuroimaging markers using amyloid PET will provide more specific outcome measurements of vascular amyloid elimination. This may be made possible through the development of PET tracers, such as the Ga-P14-032 PET probe, that have increased specificity to bind to vascular amyloid compared to traditional tracers [277].
Certain limitations need to be addressed in CAA studies to provide more comprehensive information on CAA and its downstream paths. For example, many studies have limited statistical power due to small sample sizes and have focused on cross-sectional design restricting interpretations about CAA progression. Additionally, studies could benefit from incorporating a range of biofluid markers, cognitive assessments, and neuroimaging measures to inform clinicians and scientists of markers that can be used to identify brain vulnerability related to CAA. Collaborative efforts in big data studies may help amplify and accelerate the search and development of therapies to prevent and/or manage CAA and reduce AD prevalence.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgements to report.
FUNDING
This review was funded by the NIH grant U19AG078109.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.