
Abstract
A computational study about the effect of BX3 (X = H, F, Cl and Br) interaction in C–H acidity enhancement of some aldehyde, ketone and imine molecules is performed by B3LYP/6- 311++G(d,p) method in gas phase. The boron derivatives of model molecules show more acidity in comparison with their pure forms. This acidity improvement is attributed to the effective interaction of the C = O/C = N group with the B atom of BX3. The acidity enhancement is according to the BBr3 > BCl3 > BF3 > BH3 order which shows that boron compounds with electron withdrawing groups and especially BBr3 can be used as an effective and promising C–H activator in various organic reactions.
Introduction
The C–H bond activation is defined as the direct functionalization of a non-acidic C–H bond [1–4]. This technique provides the possibility of use from the numerous inexpensive starting materials for the production of organic molecules with complex structures. The energy arrange of C–H bond dissociation is sp > sp2 > sp3 and 1° > 2°> 3°> allylic C, according to the stability of the produced radicals via hemolytic bond dissociation. Moreover, the stability of the deprotonated species affects and except of the allyl C–H bond, the pKa values tendency goes almost in the reverse orientation [5–7].
The first intention of C–H activation process is the probability of methane transformation to beneficial oxidation products i.e. methanol. By expanding the industrial techniques such as hydrofracking which increased the worldwide supply of methane, C–H activation and methane conversion have attracted more considerations [8]. The C–H activation moved forward by recognizing of catalytic role of palladium for sp3 and sp2 C–H activation in coupling reactions [9, 10]. However, due to the high prices of palladium, its application always is restricted and if catalytic activities allows, cheaper metals (or metalloids) are always of interest. For example, Shang et al., investigated the catalytic role of Fe in C–H bond activation [11]. Recently, the organocatalysts have been attracted great attention in C–H bond activation because of their green and safe nature [12]. However, exploring the novel, safe and effective reagents for C–H bond activations still remains as an interesting research field.
The acidity enhancement in boron compounds have been attracted more attention in both theoretical and experimental studies in last decade [13–17]. Alkorta et al., [18] investigated the bonding in complexes X:BH3- n F n and X:BH3- n Cl n for X = N2, HCN, LiCN, H2CNH, NF3 and NH3 with n = 0–3. They found that the interaction energies of mentioned complexes are controlled by three factors; the reduce in the electron-accepting nature of boron atom as a result of π donation by F or Cl atom, the increase in the electron-accepting nature of boron atom because of acid deformation and big increase in the deformation energy of the acid due to the increasing halogen substitution. Hurtado et al., [19] found that the acidity of BH3 in gas phase phosphine-boranes is 13 and 18 orders of magnitude in terms of ionization constants compared to the corresponding free phosphines. In the other work, Hurtado et al. [20] compared the intrinsic acidity of cyclopenta-2,4-dienylborane and its Al and Ga analogues, computationally. They found that the substitution of one of H atoms of the C(sp3)H2 group of cyclopentadiene by an XH2 (X = B, Al, Ga) causes to an improvement which is considerably large for boron derivative.
The computational chemistry gives obvious and useful vision into the mechanisms of chemical reactions. Numerous papers and review articles have been published about computational aspects of C–H bond activation [21–24]. The researchers have studied various aspects of C–H bond activation such as oxidative addition, reductive elimination, insertion, metathesis from computational and theoretical viewpoint [25–29].
In the continuation of previous studies [30–39] and by inspiring from the literature [40–43] we have carried out density functional theory (DFT) calculations to study the interaction between some boron compounds such as BH3, BF3, BCl3 and BBr3 and some model aldehyde, ketone and imine molecules for exploring of boron role in C–H bond activation in mentioned molecules.
Computational method
All calculations were performed by Gaussian 03 software [44]. Initially, the chemical structures of all model molecules and their corresponding conjugated bases were fully optimized at B3LYP/6–311++G(d,p) level of theory. The small basis set causes to a significant Basis Set Superposition Error (BSSE) such that, in some cases, ordinary correction by the counterpoise method entirely eliminates dispersion bonding [45]. Due to the fact that the reduction of BSSE by using larger basis sets is preferable to applying counterpoise corrections [45], 6–311++G(d,p) basis set was applied for all calculation. The vibrational frequencies were further calculated at the same level to characterize that each optimized kind was a minimum on the potential energy surface. The local minima on the potential energy surface have no negative eigenvalue. In order to study the acidity improvement of the proposed molecules, all the side parameters such as self-association, solvent, temperature etc. (which may alter the most stable conformation) were ignored and only single molecule was optimized at the mentioned level of theory and investigated in the gas phase. The most stable conformers of the molecules e.g. propanal and 3,3,3-trifluoropropanal was checked by the literature [46]. Ethanal (1), propanal (2), 3,3,3-trifluoropropanal (3), heptanal (4), cyclopropanone (5), cyclobutanone (5′), cyclopentanone (5′′), cyclohexanone (5′′′), ethanimine (6), propan-1-imine (7), 3,3,3-trifluoropropan-1-imine (8), heptan-1-imine (9), cycloprpanimine (10), cyclobutanimine (10′), cyclopentanimine (10′′) and cyclohexanimine (10′′′) were selected as model molecules (Fig. 1). The thermodynamic quantities such as enthalpy and Gibbs free energy for deprotonation reaction in gas phase (ΔHacid and ΔGacid) were obtained via the frequency calculations at B3LYP/6–311++G(d,p) level of theory and 298 K using Equations 2:
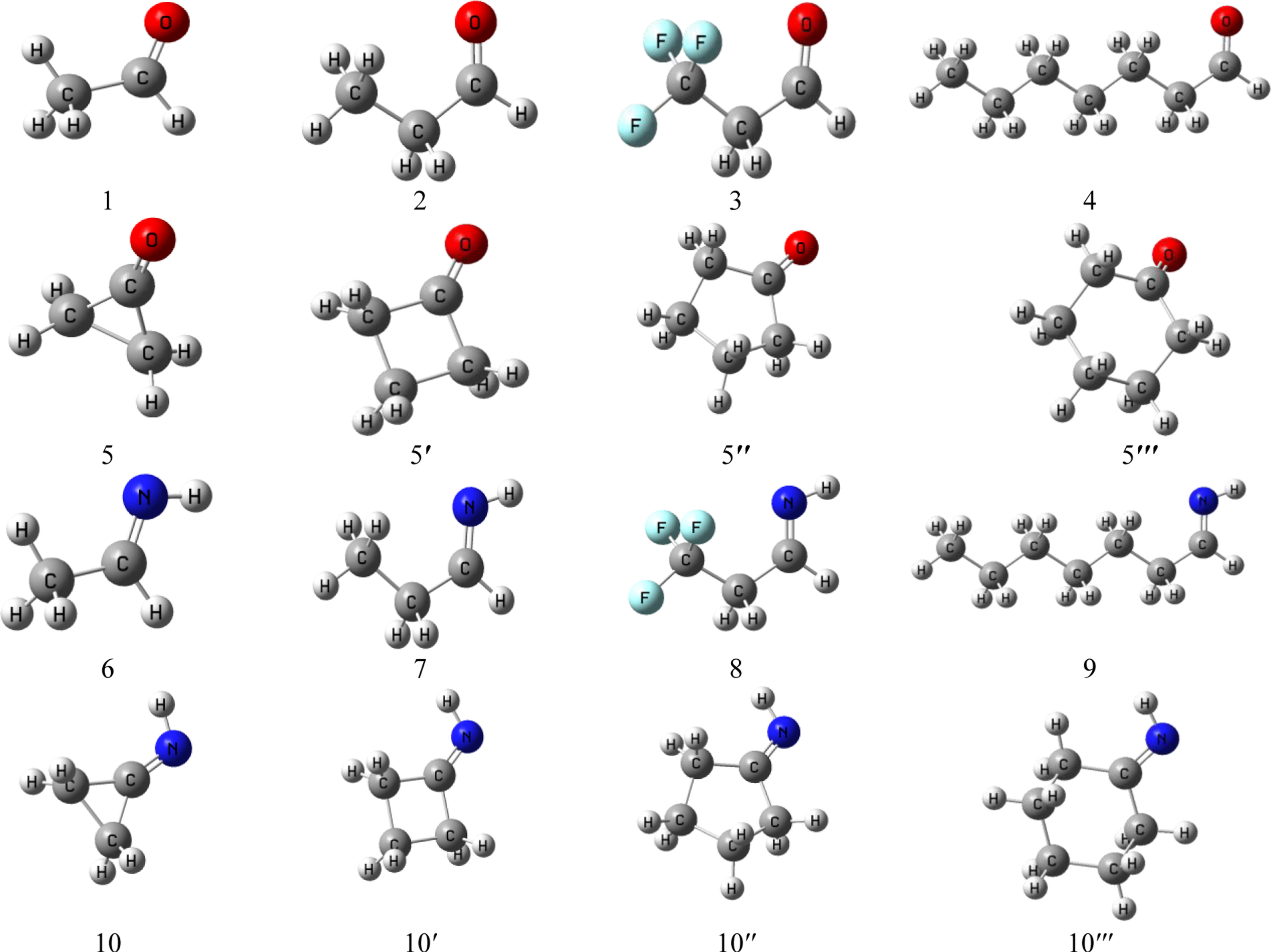
Optimized structures of model molecules at B3LYP/6–311++G(d,p) level of theory.
In above equations, H(H+) and G(H+) are +0.00236 and –0.01 hartree, respectively. Since, both the ΔHacid and ΔGacid are generally positive amounts; their smaller values demonstrate more acidity. In order to benchmark the selected B3LYP/6–311++G(d,p) level of theory, four levels of theory including B3LYP/6–31G(d), B3LYP/6–311++G(d,p), MP2/6–31G(d) and MP2/6–311++G(d,p) were used for calculation of ΔHacid of ethanal molecule. The calculated values were obtained as 1519.8, 1524.3, 1525.7 and 1527.5 kJ mol–1. respectively. Mead et. al., [47] reported that the ΔHacid for ethanal is 1533.1±3.4 kJ mol–1 in gas phase. It can be seen that the results obtained from MP2 method are closer to the experimentally obtained result than that of B3LYP. Moreover, the reality of the results with 6–311++G(d,p) basis set is more than 6–31G(d). However, due to the time-consuming characteristics of MP2 method, its high computational cost, a little difference between MP2 and B3LYP results and also according to the previous studies which proved the B3LYP/6–311++G(d,p) level of theory will give consistently reliable geometries, conformations and energies for common simple molecules [48], this level was selected as an appropriate level of theory for further calculations.
The optimized chemical structures of some model aldehyde, ketone and imine molecules with borane derivatives (BX3; X = H, F, Cl, Br) are shown in Fig. 2. In compounds 1a-1d, the effect of X type of BX3 molecule in activation of the selected C–H bond in ethanal has been studied. For exploring the generality of the results, the same computations were performed on propanal (2a-2d). The effect of an electron withdrawing group (CF3) in the adjacent position of C–H bond was studied in 3,3,3-trifluoropropanal as a model compound 3a-3d. The heptanal was selected as a model compound in order to study the effect of O . . . BF3 interaction on activation of the various C–H bonds in the molecule (4a). The activation of C–H bonds in ketones was investigated by cyclopropanone, cyclobutanone, cyclopentanone and cyclohexanone in 5a, 5′a, 5′′a and 5′′′a, respectively. In order to study the effect of borane derivatives in activation of C–H bond in imines, ethanimine (6a-6d) and propan-1-imine (7a-7d) were selected. Also, the effect of adjacent CF3 group in activation of C–H bond was investigated using 3,3,3-trifluoropropan-1-imine as a model compound (8a-8d). For studying the effect of BF3 in activation of the different C–H bonds in long chain imines, heptan-1-imine was selected (9a). Finally, the effect of BF3 in the activation of C–H bond in cyclic imines was studied using cycloprpanimine, cyclobutanimine, cyclopentanimine and cyclohexanimine in 10a, 10′a, 10′′a and 10′′′a, respectively.
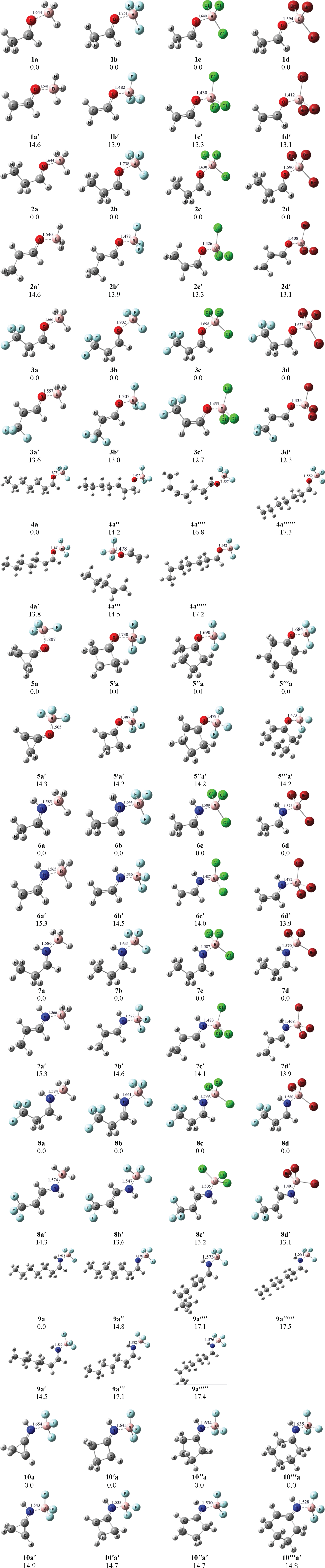
Optimized structures and relatives energies for the isomers of the borane (-BX3) derivatives of model molecules at B3LYP/6–311++G(d,p) level of theory. The bond lengths and energies are in Å and kJ mol–1, respectively.
In order to improve interaction strength of borane with model molecules, its hydrogen atoms were replaced by some electron withdrawing groups such as F, Cl and Br atoms. The relative energies demonstrate that the deprotonated compounds in each case are more stable than neutral ones because deprotonated structures contain a negative charge which easily can be imported to the vacant orbital of boron atom. The fewer O . . . BX3/N . . . BX3 interaction length in deprotonated compounds in comparison of neutral compounds verifies this claim (Fig. 2).
The calculated ΔHform and ΔGform values for the borane derivatives of variety model molecules are summarized in Table 1. It should be said that the calculated values are relevant to deprotonation of the most stable neutral model molecules and hence formation of the most stable relevant deprotonated ones. Table 2 represents the ΔHacid and ΔGacid values for deprotonation of pure and boron complex forms of the model molecules. The comparison of the enthalpy and Gibbs free energies for neutral and deprotonated ethanal show that only the ethanal complex production with BH3 is favored, thermodynamically (ΔGform = –6.41 kJ mol–1), however, the formation of complex with all boron derivatives for deprotonated ethanal are favored, thermodynamically which shows these complexes can be formed easily (Table 1, see ΔGform values for 1a′, 1b′, 1c′ and 1d′). The ΔHacid and ΔGacid values for deprotonation process of the ethanal and its boron complexes are shown in Table 2. It can be seen that the ΔGacid value for ethanal deprotonation is 1524.3 kJ mol–1. The complex formation with boron derivatives, increases the acidity of ethanal so that all the ethanal . . . BX3 complexes have lower ΔHacid and ΔGacid values than pure ethanal (Table 2, see 1a, 1b, 1c and 1d). The acidity increase depends on type of atoms which are attached to the B atom. The acidity increase is higher with BBr3 (ΔHacid = 1243.0 kJ mol–1 and ΔGacid = 1211.2 kJ mol–1) in comparison to BH3 (ΔHacid = 1379.1 kJ mol–1 and ΔGacid = 1347.8 kJ mol–1) due to the electronegativity of Br atoms.
The calculated ΔHform and ΔGform values for the borane derivatives of variety model molecules at B3LYP/6–311++G(d,p) level of theory
The calculated ΔHacid and ΔGacid values for the pure and borane derivatives of variety model molecules at B3LYP/6–311++ G(d,p) level of theory
Figure 1 demonstrates the interaction of the neutral and deprotonated propanal with boron derivatives. It can be seen that the O . . . B interaction distance in deprotonated propanal is lower than corresponding interval in neutral propanal. Though the formation enthalpies are negative for all propanal . . . BX3 complexes (Table 1, see ΔHform values for 2a, 2b, 2c and 2d), only formation Gibbs free energy is negative for propanal . . . BH3 complex (–7.16 kJ mol–1) which means that only formation of this complex is favored, thermodynamically. The interaction of anionic propanal with boron derivatives is exothermic and the formed complexes are stable from the thermodynamically viewpoint (Table 1, see ΔHform and ΔGform values for 2a′, 2b′, 2c′ and 2d′). The formed complexes with BBr3 are more stable than others because Br electron with-drawing groups are more effective in distribution of negative charge. Table 2 shows that the elimination of the α-proton in propanal is not easily executable (1528.2 kJ mol–1), however when propanal . . . BX3 complexes are formed, both ΔHacid and ΔGacid are decreased, considerably. It can be seen that the BBr3 increases the acidity of α-proton more successfully (ΔHacid = 1242.5 kJ mol–1, ΔGacid = 1211.9 kJ mol–1).
In order to investigate the effect of the presence of the electron with-drawing group on the interaction of boron derivatives with carbonyl compounds, 3,3,3-trifluoropropanal was studied. Figure 1 represents the optimized structures of the neutral and deprotonated 3,3,3-trifluoropropanal complexes with BH3, BF3, BCl3 and BBr3 molecules. The ΔHform and ΔGform values for interaction of BX3 with both neutral and deprotonated 3,3,3-trifluoropropanal are summarized in Table 1. It can be seen that only the interaction of neutral 3,3,3-trifluoropropanal with BH3 and BF3 are exothermic (–41.3 and –20.5 kJ mol–1, respectively). In addition, the ΔGform value shows that the complex formation of the neutral 3,3,3-trifluoropropanal with boron derivatives is not possible, thermodynamically. In contrast, the formation of complexes between deprotonated 3,3,3-trifluoropropanal with boron derivatives are both exothermic and favored thermodynamically (Table 1, see 3a′, 3b′, 3c′ and 3d′). The comparison of the obtained results for propanal and 3,3,3-trifluoropropanal in Table 1 show that the presence of an electron with-drawing group such as CF3 leads to weakening the interaction between carbonyl group and boron derivatives. The calculated values for ΔHacid and ΔGacid of the neutral and deprotonated 3,3,3-trifluoropropanal are summarized in Table 2 (see 3, 3a, 3b, 3c and 3d). It can be concluded that CF3 group increases acidity of the α-proton so that, the acidity of α-proton in the 3,3,3-trifluoropropanal (ΔHacid = 1420.8 kJ mol–1) is about 100 kJ mol–1 higher than propanal (ΔHacid = 1528.2 kJ mol–1). Moreover, the acidity of the α-proton for 3,3,3-trifluoropropanal has been increased about 200–300 kJ mol–1 in the presence of boron derivatives so that, 3,3,3-trifluoropropanal complex with BBr3 with ΔHacid = 1170.9 kJ mol–1 shows higher acidity than citric acid.
For exploring that how boron interaction effects on activation of other C–H bonds that they are far away from carbonyl bond, we selected heptanal molecule. The optimized structure for BF3 complex of heptanal and its deprotonated structures from various C–H bonds are shown in Fig. 2 Table 1 shows that the complex formation with BF3 for heptanal is exothermic (–40.3 kJ mol–1), however, due to its positive ΔGform value (13.9 kJ mol–1), this interaction is not favored, thermodynamically. In the other word, heptanal . . . BF3 complex is not stable, however, the ΔHform and ΔGform values of deprotonated heptanal with BF3 show that the formation of these complexes are favored, thermodynamically and they are stable. The main reason for stronger interaction of BF3 with deprotonated heptanal molecules is the presence of negative charge which helps oxygen atom of carbonyl group gives its lone pair to the vacant orbital of boron atom, more easily. Furthermore, comparison of ΔHform values show that the BF3 interaction with carbonyl weakens when the anionic center goes away from carbonyl group, however this trend does not decrease, regularly. The ΔHacid and ΔGacid values for deprotonation of heptanal from different sites are shown in Table 2. It can be seen that the proton removing from α-position is easier than further positions (ΔHacid = 1512.4, 1684.9, 1687.9, 1677.3, 1711.5 and 1711.5 kJ mol–1 for removing α, β, γ, δ, ɛ and ζ-proton, respectively). Moreover, it is obvious that the complex formation with BF3 leads to increase acidity of heptanal carbon atoms, however this enhancement is different for various carbon atoms. The haptanal . . . BF3 complex increases the acidity of α-carbon about 200 kJ mol–1, however, further carbon atoms from carbonyl group experience lower acidity enhancement so that, the ζ carbon atom shows about 80 kJ mol–1 acidity increase.
In the next step, some of cyclic ketones such as cyclopropanone, cyclobutanone, cyclopentanone and cyclohexanone were considered for investigating of C–H bond activation in ketone molecules. The optimized chemical structures of mentioned ketones are shown in Fig. 1. The comparison of O . . . B interaction distance in neutral and deprotonated ketones reveals that the interaction of BF3 with deprotonated ketones are stronger than neutral ones. The enthalpy and Gibbs free energy values for interaction of BF3 with mentioned cyclic ketones are summarized in Table 1. The ΔHform values show that increasing the ring size leads to enhancing O . . . B interaction (see ΔHform values for 5a, 5b, 5c and 5d), however complex formation of BF3 and these ketones are not favored, thermodynamically and these complexes are not stable. In contrast, the deprotonated cyclic ketones can form stable complexes with BF3 molecule which their formation is favored, thermodynamically (Table 1, see ΔGform values for 5a′, 5b′, 5c′ and 5d′). Again, the ΔHform and ΔGform values for O . . . B interaction in deprotonated ketones increase in larger rings which means that the large cyclic ketones can form more stable complexes with BF3. The calculated ΔHacid and ΔGacid values for deprotonation of the α-carbon in mentioned ketones and their BF3 complexes are summarized in Table 2. The comparison of the ΔHacid values reveals that the acidity property of their α-protons are almost equal (1534.5, 1528.3, 1532.4 and 1536.3 kJ mol–1) and cyclobutanone shows a bit more acidity than others. The complex formation with BF3 also enhances the acidity of α-carbon in the ketone molecules about 200 kJ mol–1. The acidity of α-carbons in the BF3 complex of the cyclic ketones does not differ with each other, significantly, however, cyclobutanone complex with BF3 is shows more acidity than others (ΔHacid = 1340.9 kJ mol–1). As cyclopentanone and cyclohexanone have two different kinds of α-protons, the deprotonation of every two types was studied. The obtained results did not show the considerable deference between ΔHacid values for two α-proton types of cyclopentanone complex with BF3 (1343.8 and 1342.1 kJ mol–1), however, in the complex of cyclohexanone with BF3, a bit more difference is observed between deprotonation of axial and equatorial protons (1342.2 and 1339.4 kJ mol–1, respectively). This phenomenon can be attributed to the existence of 1,3-steric interactions in axial position and the molecule tendency for releasing from this steric by deprotonation.
After the investigating the interaction of various boron derivatives with carbonyl groups and subsequent C–H activation in aldehydes and ketones, we studied their interaction with imine functional group and its effect on acidity enhancement. The optimized structures of neutral and deprotonated ethanimine with BH3, BF3, BCl3, BBr3 are shown in Fig. 1. The comparison of N . . . B interaction distance between complexes of neutral and deprotonated ethanimine with BX3 molecules demonstrate that this interaction is stronger in depronotated imine than neutral molecule. The calculated ΔHform and ΔGform for complexes of ethanimine and BX3 molecules show the stability of the formed ethanimine complexes with BX3 (Table 1). Moreover, it can be seen that the interaction of the deprotonated ethanimine with BX3 compounds are stronger than neutral ethanimine which can be attributed to the existence of the localized negative charge which helps the nitrogen atom to give its lone pair to boron’s vacant orbital. Table 2 shows the ΔHacid and ΔGacid values for deprotonation of the α-proton in the ethanimine and its BX3 complexes. The ΔHacid value for ethanimine is 1580.9 kJ mol–1 which demonstrates its weak acidity. The comparison of ΔHacid of ethanal and ethanimine shows that the acidity of α-carbon in ethanimine is higher than ethanal. In addition, the acidity of the α-carbon has been increased about 200–280 kJ mol–1 after formation the complex with BX3 molecules.
The effect of the methyl group on the interaction of boron compounds and imines was investigated. So, propan-1-imine was selected as a model compound. The optimized structures for propan-1-imine and its complexes with BH3, BF3, BCl3 and BBr3 are shown in Fig. 1. The ΔHform and ΔGform values were calculated for complexes and it can be seen that their formation are exothermic and all of them are stable, thermodynamically (Table 1, see the ΔHform and ΔGform values for 7a, 7b, 7c and 7d). The comparison of ΔHform and ΔGform values for ethanimine and propan-1-imine reveals that the addition of a methyl group to imines facilitates the electron transfer from nitrogen atom to boron’s vacant orbital and strengthens the N . . . B interaction due to the methyl’s electron donating characteristic. The calculated ΔHform and ΔGform values show that the interaction of the boron compounds with deprotonated ethan-1-imine is higher than neutral molecule (Table 1). The comparison of ΔHacid and ΔGacid of ethanimine and propan-1-imine reveals that the acidity of the α-carbon decreases about 3 kJ mol–1 in propan-1-imine because of the methyl’s electron donating property (Table 2). It can be seen that the interaction of boron compounds with propan-1-imine increases the acidity of the α-carbon. This enhancement depends to the BX3 type of molecule so that, the most increase can be seen in the presence of BBr3 which elevates the acidity about 280 kJ mol–1.
The CF3 group is a strong electron with-drawing group and it is expected that affects the imines acidity. So, the interaction of 3,3,3-trifluoropropan-1-imine and its deprotonated form was investigated with BX3 molecules (Fig. 2). The comparison of the N . . . B interaction distances show that deprotonated 3,3,3-trifluoropropan-1-imine can make stronger interaction with BX3 molecules than neutral one (Table 1). The ΔHform and ΔGfrom values for 3,3,3-trifluoropropan-1-imine demonstrate that the formation of all these complexes are exothermic and favored, thermodynamically (Table 1). The comparison of the ΔHform values for propan-1-imine and 3,3,3-trifluoropropan-1-imine represents that the addition of CF3 group decreases N . . . B interaction. Moreover, the interaction of BX3 compounds with deprotonated 3,3,3-trifluoropropan-1-imine is higher than neutral molecule (∼150 kJ mol–1). Also, as charge distribution characteristics increases from BH3 to BBr3, it can be seen that the interaction of deprotonated 3,3,3-trifluoropropan-1-imine with BX3 enhances according to BBr3 > BCl3 > BF3 > BH3 order (Table 1, see ΔHform values for 8a′ to 8d′). Table 2 shows that the addition of a CF3 group leads to increase of the α-carbon acidity in imines so that, 3,3,3-trifluoropropan-1-imine represents about 120 kJ mol–1 higher acidity than propan-1-imine. The formation of complex with BX3 compounds increases the acidity of 3,3,3-trifluoropropan-1-imine, however, BBr3 has most considerable effect on the acidity (ΔHacid = 1241.7 kJ mol–1).
For investigating the boron compounds effect on the acidity of various carbon atoms in the imines, heptan-1-imine was selected as a model molecule (Fig. 1). Table 1 shows that ΔHform and ΔGform for the complexes of neutral and deprotonated forms of heptan-1-imine with BF3 molecule (9a to 9a′′′′′′). It can be seen that all the interactions are exothermic and the formed complexes are stable, thermodynamically. As expected, the interaction of anionic heptan-1-imine with BF3 was stronger than neutral molecule (see ΔHform values for 9a to 9a′′′′′′). Moreover, the interaction strength with BF3 depends to the carbon which loses its hydrogen atom so that, the ΔHform decreases as anionic center goes away from N . . . B interaction canter. Table 2 demonstrates that the acidity of carbon atoms decreases in those are far from iminic functional group, so that the ΔHacid value are calculated 1563.5 kJ mol–1 and 1721.7 kJ mol–1 for α and ζ carbons, respectively. The complex formation of heptan-1-imine with BF3 molecule increases the acidity of all carbon atoms, however, this enhancement is more for closer carbon atoms to the imine functional group.
In order to exploring the activation of C–H bond in cyclic imines with BF3 molecule, cyclopropanimine, cyclobutanimine, cyclopentanimine and cyclohexanimine were selected as model compounds. The optimized structures for neutral and deprotonated forms of these imines are shown in Fig. 2. The calculated ΔHform values for cyclopropanimine and cyclohexanimine are –83.2 kJ mol–1 and –226.8 kJ mol–1, respectively (Table 1), which can be attributed to the larger ring size in cyclohexanimine and presence of more CH2 electron donating groups in the molecule which facilitate electron transfer from anionic carbon atom to the vacant boron’s orbital and thus, strengthening the N . . . B interaction. The ΔHacid and ΔGacid values reveal that with increasing the ring size, the acidity of the α-carbon in cyclic imines remains almost fixed with little difference (Table 2). The complex formation with BF3 increases the acidity of these cyclic imines about 200 kJ mol–1. Similar to cyclopentanone and cyclohexanone, the acidity of two kinds of α-protons in cyclopentanimine and cyclohexanimine in BF3 complexes were investigated. There is no significant difference between deprotonation of two types of hydrogens in the cyclopentanimine molecule (1392.9 and 1392.1 kJ mol–1). In the cyclohexanimine complex with BF3, the difference was a little more due to the 1,3-diaxial steric effects and the axial proton was a bit more acidic than equatorial one (1397.7 and 1395.9 kJ mol–1, respectively).
The interaction of BX3 species with the selected molecules can be investigated by the charge transfer value. The natural bond orbital (NBO) and Mulliken charge analysis are useful analyses for investigating the intermolecular interactions, and moreover are appropriate method for study the intermolecular net charge transferring [49, 50]. In the NBO analysis, the energy differentiation between bonding and anti-bonding orbitals makes the molecule potent for interactions. The biggest difference between NBO and Mulliken analyses is that the NBO is considerably less basis set dependent than Mulliken populations [49]. So, the NBO analysis was applied for ethanal as the sample molecule in this section at B3LYP/6–311++G(d,p) level of theory (Table 3). There are no considerable changes in the electronic characteristics of deprotonated ethanal complexes with BX3 molecules (Table 3; 1a′, 1b′, 1c′ and 1d′). However, during interaction of ethanal with mentioned spices, the considerable changes in the electronic properties were observed (Table 3; 1a, 1b, 1c and 1d). The obtained results demonstrated that amount of charge transfer from the oxygen atom of the ethanal to the boron atom of the BX3 species increases according to the BBr3 > BCl3 > BF3 > BH3 order. These results are totally in agreement with the results of ΔHform and ΔGform values and can attributed to the better BBr3 ability to disturbing the negative charge in comparison with BCl3, BF3 and BH3 molecules.
The natural bond orbital (NBO) analysis of natural and deprotonated ethanal with BH3, BF3, BCl3 and BBr3 at B3LYP/6–311++G(d,p) level of theory
A computational study was performed on the interaction effect of boron compounds involving BH3, BF3, BCl3 and BBr3 on C–H activation in some aldehydes, ketones and imines via calculation of ΔHform/acid and ΔGform/acid values. The boron compounds are flat, however, after the interaction with carbonyl compounds, they turn to be in semi-pyramidal structure. The presence of electron with-drawing groups such as F, Cl and Br causes to increasing the interaction strength between target compounds and BX3 molecules. The ΔHform values showed that the interaction of BX3 molecules with deprotonated model compounds are very stronger than that neutral ones and also according to the ΔGform values it can be said that they are spontaneous (ΔGform < 0). When a CF3 group was added to the model molecule, ΔHform values were decreased, because CF3 is a strong electron with-drawing group. So, the electron transfers from molecule to boron’s vacant orbital and therefore molecule . . . BX3 interaction becomes difficult. In the cyclic ketones and imines, the ΔHform and ΔGform values increase with increasing the ring sizes due to the electron donating roles of CH2 groups which reinforces electron transfer from carbonyl to the boron’s vacant orbital. The main reason for the better interaction of BBr3 and carbonyl or iminic groups is its ability to disturbing the negative charge in comparison to BH3, BF3 and BCl3. The obtained results showed that all the boron compounds increase the acidity of target molecules and BBr3 and BH3 have the most and least effects on the acidity enhancement, respectively. The CH3 group does not have a considerable effect on the carbon acidity in the complex form, even it causes to decrease the acidity in some cases. In contrast, the CF3 group has a significant effect on the acidity increase so that, ΔHacid value of 3,3,3-trifluoropropanal is higher than acetic acid. In long chain aldehyde and imine molecules, the acidity of hydrogen atoms decreases by getting away from C = O . . . BX3 or C = N . . . BX3 groups. Finally, it can be said that the acidity enhancement of the target compounds with BX3 molecules is according to the BBr3 > BCl3 > BF3 > BH3 order and boron compounds can be used as effective mediator for activation of C–H bond in organic synthesis.
Footnotes
Acknowledgment
The authors thank Islamic Azad University, Science and Research Branch for the partial support.
Ethical statement
We have no conflicts of interest to declare.