
Abstract
Favipiravir (Fav) has become a well-known drug for medication of patients by appearance of COVID-19. Heterocyclic structure and connected peptide group could make changes for Fav yielding different features from those required features. Therefore, it is indeed a challenging task to prepare a Fav compound with specific features of desired function. In this work, existence of eight Fav structures by tautomeric formations and peptide group rotations were obtained using density functional theory (DFT) optimization calculations. Gas phase, octanol solution, and water solution were employed to show impact of solution on features of Fav besides obtaining partition coefficients (LogP) for Fav compounds. Significant impacts of solutions were seen on features of Fav with the obtained LogP order: Fav-7 > Fav-8 > Fav-4 > Fav-3 > Fav-2 > Fav-5 > Fav-1 > Fav-6. As a consequence, internal changes yielded significant impacts on features of Fav affirming its carful medication of COVID-19 patients.
Introduction
By the end of 2019, coronavirus disease (COVID-19) has been spread all around the world with serious harmful effects on human health and life systems [1–3]. Although it was a member of already known coronaviruses, but the new functions made it a wild virus banning the life from regular cycle [4–6]. Therefore, several attempts have been dedicated to explore a way of medication for the infected patients in addition to prevention protocols and lock down [7–9]. Among such attempts, exploring effects of available drugs on the mechanism of disease has been a quick trial for exploring a useful drug for medication of COVID-19 patients [10–12]. Favipirvir (Fav) has been one of those drugs with expectation of medication of COVID-19 patients from early days of appurtenance of disease [13–15]. Although it was available, but considerable efforts have been carried out to show various features of this drug especially regarding the chemical structure properties [16–18]. Moreover, its function against typical enzymes associated with coronavirus was examined by means of experiments and computations [19–21]. Hence, it has become a hot topic of research for those scientists dealing with modifying structures for achieving better efficacy for the targeted drugs [22–24]. One of the important features for chemical compounds is their solubility in octanol and water solutions known as partition coefficient (LogP) for medical chemists [25–27]. Knowing LogP for a pharmaceutical compound could help to predict its function for participating in interaction with other substances even penetrating through membranes and tissues inside the body [28–30]. It could be mentioned here that the phases and media of materials are important to define their functions even in inorganic based materials in solid phases and so on [31, 32]. Therefore, it is an important task to explore LogP for Fav to provide more insightful information on its chemistry for moving forward further related investigations [33–35]. To this aim, this work was performed to investigate chemistry of Fav in octanol/water solutions to provide LogP for this important pharmaceutical compound. To achieve this purpose, possible structures of Fav were investigated based on movement of hydrogen atom between amine and keto groups of heterocyclic structure in addition to change of angle of attached peptide group. In this regard, eight structures were evaluated to be investigated carefully in a systematic way by performing high-level quantum chemical calculations [36–38]. It is worth noting that the movement of hydrogen atom among amine and keto groups could yield formations of tautomeric structures as a regular mechanism in the heterocyclic organic compounds [39–41]. As a consequence, computations of the investigated models were done in the separated solutions of octanol and water in addition to isolated gas phase models. Computer-based tools have been always under development especially for recognition of biological systems, in which several software programs and algorithms have been introduced up to now [42–44]. All obtained quantitative and qualitative results were summarized in Table 1 and Figs. 1–4 for discussing the goal of this work to reach a point to show chemistry features of Fav in octanol/water solution media.
Obtained features for the optimized models in different media*
Obtained features for the optimized models in different media*
*Models are shown in Figure 1. G, O, and W designate Gas phase, Octanol solution, and Water solution, respectively. ETot, ΔETot, ESol, and ΔGSol are in Kcal.mol–1. LogP is unit-less. HOMO, LUMO, and EGap are in eV. DM is in Debye. ΔETot is obtained by difference of total energies of each compound from the lowest energy compound in each media. ESol is obtained by difference of total energies of each compound in each solution and gas phase. ΔGSol is obtained by difference of free energies of each compound in each solution and gas phase. EGap is obtained by difference of energies of HOMO and LUMO of each compound in each media.
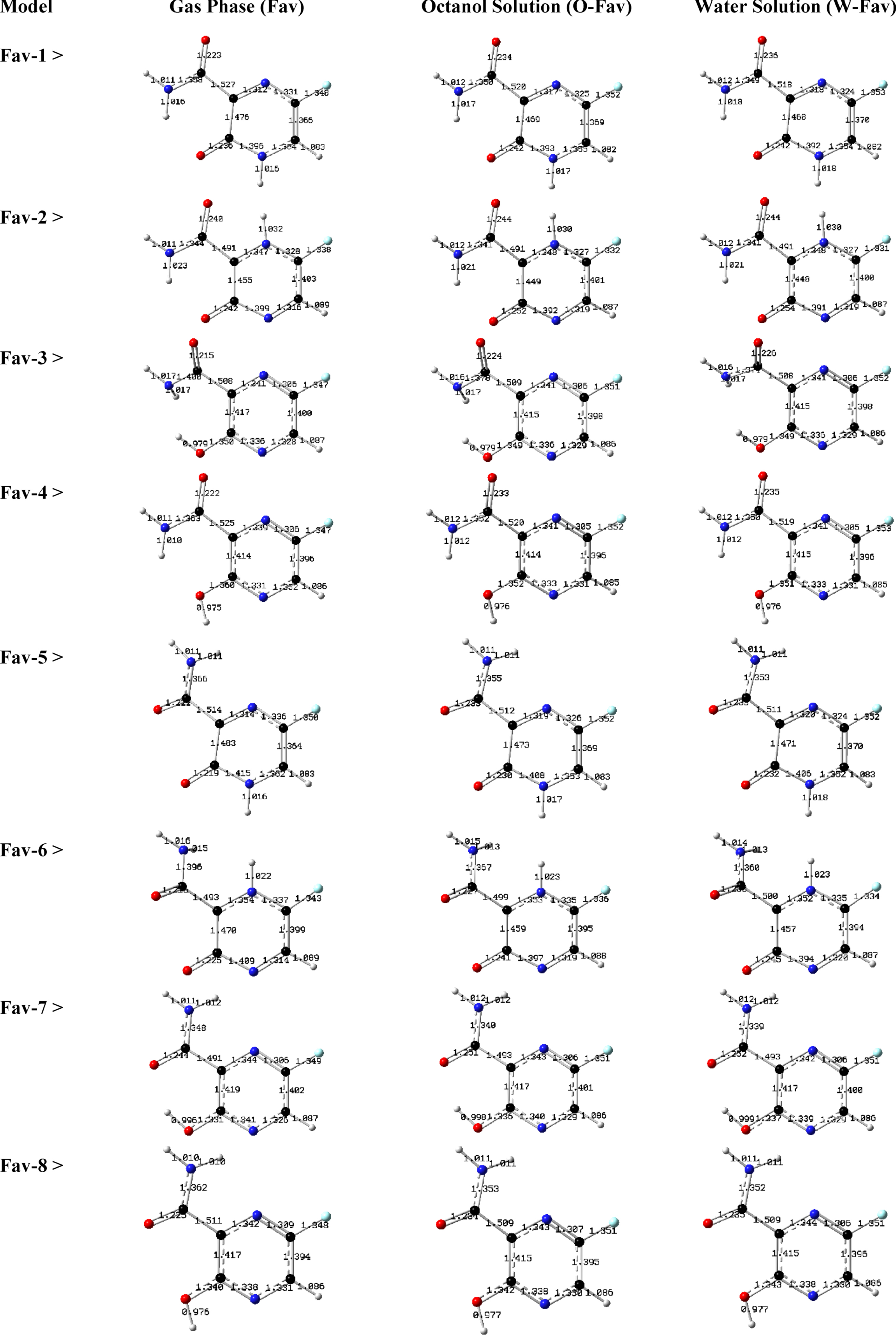
Optimized models of Fav compounds in different media. Bond distances are shown in angstrom.
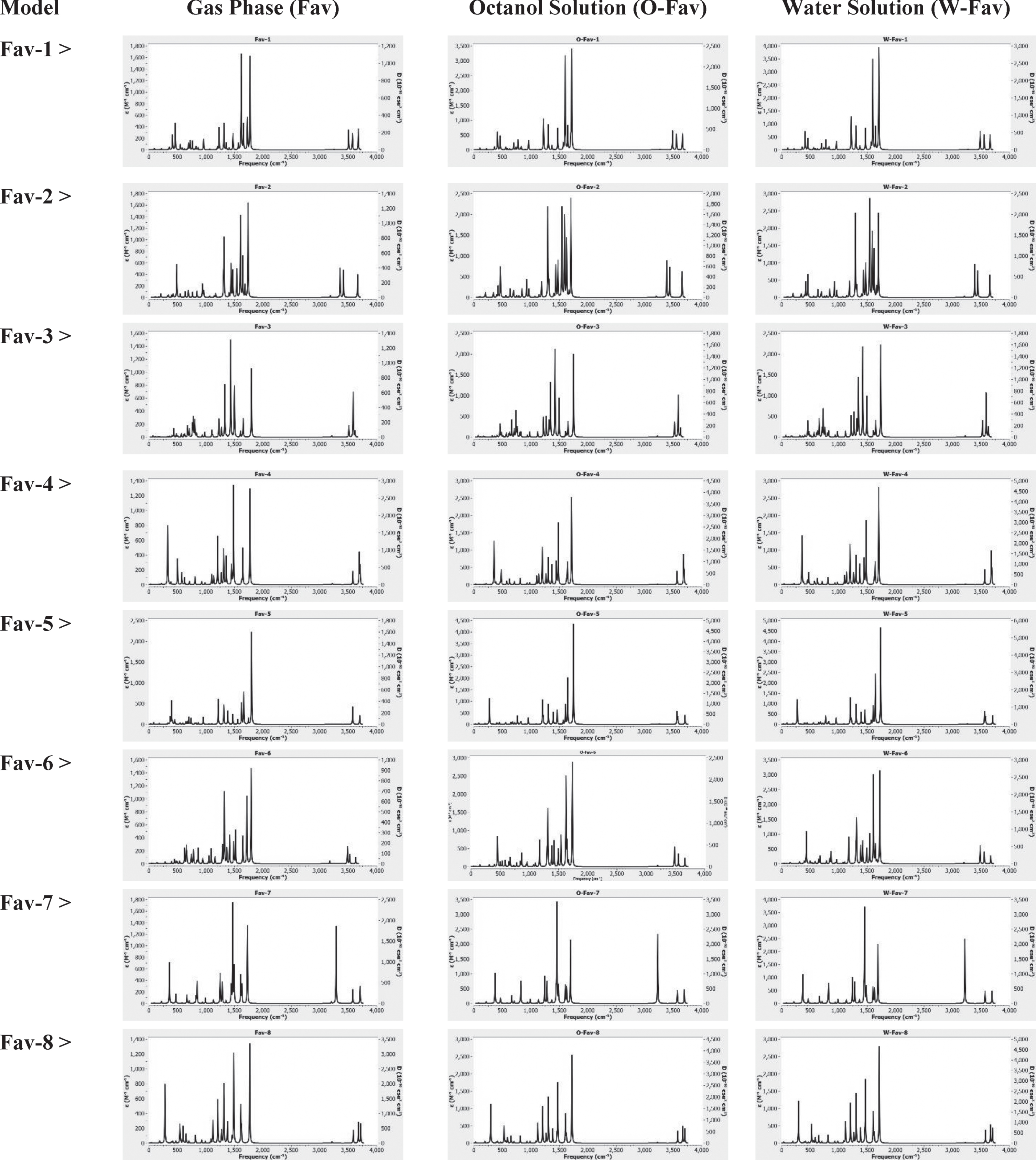
IR spectra for the optimized models of Fav compounds in different media.

NBO atomic charges for the optimized models of Fav compounds in different media.

HOMO-LUMO distribution patterns for the optimized models of Fav compounds in different media.
Within this work, 3D structures of Fav were separately optimized in isolated gas phase, octanol solution and water solution to obtained minimized energy geometries regarding the existence in different media. By movement of hydrogen atom of amine group of heterocyclic ring between nitrogen and oxygen sites, eight model structures were created for Fav (Fig. 1) for exploring chemistry features at the smallest molecular and atomic scales held by performing quantum chemical calculations. Indeed, the main goal of this work was to investigate structural features and variation of Fav in different media to yield values of LogP for them. To this aim, optimizations of structures in different media were done and the electronic features were obtained for the optimized systems. Different values of energies including total energy (ETot), solvation energy (ESol), and differences of energies (Δ) were obtained for the optimized structures. Moreover, molecular orbital features including energy levels of the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO), differences of HOMO and LUMO (EGap), and dipole moment (DM) were computed for the optimized model systems in all investigated media. For better analyzing the structures, infrared (IR) spectra were evaluated for all models as exhibited in Fig. 2. Moreover, natural bond orbital (NBO) atomic charges and distribution patterns of HOMO and LUMO of the optimized models were also exhibited in Figs. 3 4, respectively. To obtain values of LogP, thermodynamic Gibbs (G) free energies were computed to include in an equation (LogP = (ΔGOctanol solution –ΔGWater solution)/2.303RT) for obtaining the required values [45]. Temperature (T) was set to 298.15 Kelvin implying for room temperature and R, as the general constant of gases, was set to 0.00199 KCal.K–1.mol–1. All calculation were done using the B3LYP/6-31 + G* level of density functional theory (DFT) as implemented in the Gaussian program [46]. For providing solution media, polarizable continuum model (PCM) approach was employed for including each of octanol and water solution media in the calculations [47]. It is indeed an advantage of perfuming such computations for describing the features of materials at the smallest scales in a mechanistic way [48–50]. The obtained results of this work were summarized in Table 1 and Figs. 1–4.
Results and discussion
Chemistry of Fav in octanol/water solutions were investigated in this work to show structural and electronic features of this compound in various media. It was noted that the appearance of COVID-19 raised considerable attentions to develop protocols for medication of infected patients, in which Fav has been seen as a possible drug for this purpose. Therefore, performing systematic analyses could help to provide insightful information about various features of this important compound of nowadays. In addition to one original heterocyclic compound, several other related compounds could be derived by movement of hydrogen atom of amine and keto groups for making tautomeric structures. In this regard, stabilities of tautomers could be different leading to their dominant role for occurrence of mutations in biological systems [51–53]. Therefore, it is a must to see such tautomeric formations especially in pharmaceutical compounds with importance of structure-activity relationship (SAR) aspects [54]. For Fav, such hydrogen atom movement could be possible from the original structure (Fav-1) to all other forms of tautomers. In addition to such hydrogen atom movement, rotation of dihedral angle of attached peptide group is another importance for structural characterization of Fav, in which such rotation was seen for Fav in two sets of molecules 1–4 and 5–8 as shown in Fig. 1 with up-side of keto site and down-side of keto site of peptide group. Each structure was optimized in three different media including gas phase (G, Fav), octanol solution (O, O-Fav), and water solution (W, W-Fav) to show structural and electronic features of Fav related tautomeric compounds in such media. Moreover, solubility of a compound in octanol/water solutions could be designated by LogP as an important feature of ligand based drug design procedure [55]. Indeed, measuring ratio of distribution of a compound in two hydrophobic and hydrophilic solutions could help to predict for their penetration into the tissues or their distribution in human body system regarding the concepts of rational drug design [56]. Therefore, exploring such features could help to provide information about ADMET features of drug design and development [57]. Examining content of Fig. 1 could show that the skeleton of structures of all Fav compounds were remained their covalent bonding mode and bonds disconnections were not seen for the models. This achievement could mean that other structures of Fav could be existed in addition to the original one (Fav-1); however, with different optimized geometries as could be seen by the obtained bond distance for the models. Tautomerism and bond rotation in one phase or existence of solution for each compound could both change features of targeted compound as could be seen by different values of bond distances. As a consequence, it could be expected that the corresponding energies (ETot) could show effects of such variations in the optimized geometers.
Examining values of ETot in Table 1 could show such expectation, in which Fav-7 was found to be the most stable structure among all other structures in all three media. By benefits of performing computer-based works, such features could be recognized very well avoiding the existence of so many interferes of experiments. Comparing structures of Fav-7 and Fav-8 shows that one hydrogen orientation is only different in two structures leading to such variations in stabilities. In this case, possibility of existence of internal hydrogen bond interactions could be improved regarding the formation of Fav-7 as the most stable structure. Comparing values of ΔETot could show magnitudes of variation of stability for the investigated structures, win which the weakest stability was found for Fav-6 compound in all three media. Interestingly, orders of stability were almost kept unchanged for Fav compounds in all there media with the highest stability for Fav-7 and the lowest stability for Fav-6. However, stabilities of Fav compounds were increased in each of octanol and water solution phases with higher favorability for water solution as indicated by values of ESol as a difference of energies between gas phase and each of solution phases. To this point, water solution was seen to be more favorable than octanol solution for obtaining stable compounds of Fav. Interestingly, changes of values of such solubility favorability were different for the model systems, in which the obtained values of LogP by assistance of obtained values of ΔGSol could show such achievement. Smaller values of LogP could indicate more water solubility for a structure and larger values of LogP could indicate more octanol solubility for a structure. Among the models, Fav-7 was seen to be more favorable for water solution and Fav-6 was seen to be more favorable for octanol solution as indicated before by the most and the least stabilities. The obtained values of LogP could arrange Fav compound in this order: Fav-7 > Fav-8 > Fav-4 > Fav-3 > Fav-2 > Fav-5 > Fav-1 > Fav-6, from larger to smaller values. In many cases, chemists will try to modify structures by other functional groups to obtain desired value of LogP, in which the obtained results of this work could show that the internal changes of one structure could yield several values of LogP with the same chemical formula. This achievement is also very much important regarding the dominant roles of tautomeric formations for arising mutations in biological compounds or side effects in medicinal compounds emphasizing on essence of carful characterizing all details of chemical compounds before their use in clinical trials. A drug is a useful compound, but it could be also a poison for human regarding the concept of SAR of drug design protocols; therefore, many details are required for proceeding a rational drug design protocol.
Based on obtained results of optimized geometries, energies, and LogP, variations of structural features were seen for the investigated Fav compounds in all three media. For better analyzing such variations, IR spectra of all models were shown in Fig. 2. Examining such obtained IR spectra could show different strengths of vibrations in the models systems affirming the earlier achievements on such structural recognition. The most significant changes among eight models were seen for the region around 3500 cm–1 regarding the amine of peptide group and also for the region around 1500 cm–1 regarding the keto of heterocyclic structure and that of peptide group. As a consequence, such structural features were recognized by means of obtained IR spectra for the models. Moreover, slight changes of spectra among three media were also considerable for one structure showing the impacts of solution environment for the models as designated earlier by the obtained values of LogP.
Besides such structural features, NBO atomic charges and electronic molecular orbital features were also obtained for analyzing the models based on the characteristic orbital features (Figs. 3 4). Changes of structural geometries and also impact of di-electric constants of different media yielded different atomic charges for the optimized models. Such atomic charges could provide information for future contribution of the molecular systems to interactions with other substances, in which such features could indeed predict behaviors of molecules by the roles of atomic sites in molecular interactions. It could be seen that the atoms of identical positions do not show identical charge features regarding variations of structural features in optimization processes. Since all models had the same numbers of electrons and atoms, identical numbers of molecular orbitals were available for all molecular models of Fav. However, energy levels of HOMO and LUMO were changed among the models in one phase or all three media showing the effects of structural changes and existence of solutions for yielding characteristic electronic features. Indeed, it is important to mention here that such values of energy levels of HOMO and LUMO and their differences (EGap) could imply for conductivity features of a molecule, which are important in semiconductors developments [58, 59]. Additionally, such features could be applicable for detection of existence of a specific structure regarding the importance of separation and analyzing structures especially for those tautomeric formations. Fortunately, variations of such energy levels of HOMO and LUMO in addition to the values of EGap could make possible detection of tautomeric formations for Fav compounds. Changes of environment had direct impact on levels of HOMO and LUMO for each Fav compound moving them to upper or lower levels. Moreover, such variations were obvious by comparing values of EGap. Distribution patterns of HOMO and LUMO (Fig. 4) could show slight changes of such patters for a structure in three media but showing more significant changes for 1–8 compounds of Fav. Significant observations were found for Fav-4 and Fav-8 in the gas phase, in which existence of solution made a balance for such patterns with other models. Dominant roles of structural changes and solution existence were seen for the models by variations of HOMO and LUMO distribution patterns. Values of DM also showed variations of electric charge balance in the models systems with more or less significant effects for each model/phase.
Conclusion
Chemistry of Fav in octanol/water solutions were investigated in this work regarding the importance of this compound for medication of infected patinas by COVID-19. Fav is a heterocyclic structure with a connected peptide group, in which formations of tautomers and rotations of peptide groups could both yield new features for the targeted Fav compound. Hence, eight structures were found by performing optimization processes, in which their stability in one phase or in three media; gas phase, octanol solution, and water solution, were different. In correspondence with such structural variations, obtained values of energies indicated significant impacts on stabilization energies even on solvation energies. Favorability of relaxations of molecules were seen for water solution and then in octanol solution. The results indicated such favorability by the obtained values of LogP in this order: Fav-7 > Fav-8 > Fav-4 > Fav-3 > Fav-2 > Fav-5 > Fav-1 > Fav-6 showing serious impacts of internal changes on solubility favorability of molecules. Interestingly, Fav-7 was the most stable structure and Fav-6 was the least stable structure among all eight molecules in three media. Further analyses of the models by visualized IR spectra showed significant changes of regions around 3500 cm–1 and 1500 cm–1 because of structural changes and solution existence. To this point, visualized representations of HOMO and LUMO showed slight changes of distribution patterns for one structure in three media but more significant changes for eight compounds. Existence of solution made a balance for HOMO distribution patterns of Fav-4 and Fav-8 parallel to achievements of distribution patterns of other structures. It was shown that changes of energy levels of HOMO and LUMO and their EGap could make possible the recognition of tautomeric formations or structural variations for Fav compounds. As a consequence, different features were seen for Fav compound warning the scientists to be more careful about synthesizing this compound and using it for medication of COVID-19 patients.