
Abstract
Background
Carotenoids, potent antioxidants in fruits and vegetables, have recently garnered attention for their potential therapeutic effects against neurodegenerative diseases. This study focuses on the interaction and anti-aggregation properties of conventional and unconventional carotenoids found in red mamey fruit, a nutraceutical fruit that is a rich source of these compounds.
Objective
To assess computational the interaction between of amyloid-β (Aβ) peptide with a set of carotenoids and three carotenoids previously explored in experimental assays as well as to assess ADMET prediction of carotenoids selected by computational analysis results.
Methods
We analyzed the interaction between these carotenoids and Aβ peptides using molecular docking, a key factor in Alzheimer's disease (AD) pathology. Selected carotenoids were compared with the reference compounds cryptocapsin (
Results
Computational analysis identified (5R,8R)-sapotexanthin-5,8-epoxide (
Conclusions
These results provide valuable insights for future in vitro studies of most potential carotenoids (19 and 26) and the development of potential therapeutic agents for AD.
Keywords
Introduction
Alzheimer's disease (AD) is a complex and challenging disorder affecting older adults 60 years and older in the world. 1 The etiology and pathology of AD are still not clear and are multifactorial.2–5 Currently, AD affects 55 million and by 2050 estimates show that 150 million people will be affected. 6 AD is clinically challenging because of a lack of biomarkers for early diagnosis and a lack of pharmaceutical treatments to clinically manage the disease. 6 The pathology of the disease includes changes in genomics stability, over-expression of amyloid-β peptide (Aβ), accumulation of misfolded proteins, metal accumulation, changes in cell signaling pathways, alterations in gene expressions, phosphorylation of tau, neurofibrillary tangles, and others.7–9 The current focus in AD pathology includes Aβ peptide aggregation and the mechanism of Aβ induced cell dysfunction leading to neurodegeneration in AD.8,9 Ongoing efforts are focused on identifying small molecules, both synthetic and natural, in modulating Aβ production through augmenting β-secretase pathway in AβPP metabolism and preventing Aβ aggregation to protect cellular dysfunction. 10 To this end, screening of novel molecules through omics approach can lead to novel therapeutics for AD. 11 Molecules from natural sources, which have unique structural properties, have become ideal molecules to evaluate as future drugs for AD as these products often contain neuroprotective properties. 12 There are studies on the role of natural molecules as curcumin, carotenoids and others in the prevention of AD related pathology.3,13–18 These natural molecules have issues with reference to solubility and blood-brain-barrier penetration, but have very high protective activity for cell survival, with less side effects than other molecules. 18 In particular, carotenoids have been identified as natural products that can inhibit neurodegenerative pathways.19–21 As they are lipophilic, most carotenoids can cross the blood‒brain barrier. 22 Over 750 carotenoids have been found, but the anti-aggregation activity of only a few has been explored.19,22 Carotenoids generally contain 40 carbons (C40) and are composed of two terminal members (A and A’) connected by a conjugated long chain (B) (Figure 1). Carotenoids can be classified as carotenes and xanthophylls, which can be acyclic or cyclic.23–25

Schematic representation of a carotenoid.
While carotenes only contain carbon and hydrogen, xanthophylls consist of carbon, hydrogen, and oxygen; however, there are few exceptions, including apocarotenoids (containing oxygen and fewer than C40) and acyclic carotene (without oxygen and C40). Most carotenoids share a common beta-ionone ring on both sides (A and A’) of the molecule, as seen in β-carotene. Unconventional carotenoids also contain a keto-kappa group on one or both sites, such as cryptocapsin (
In the present investigation, we focus on novel carotenoids from red mamey fruit, as the fruit contains unconventional carotenoids with ĸ-ring end groups, such as sapotexanthin (
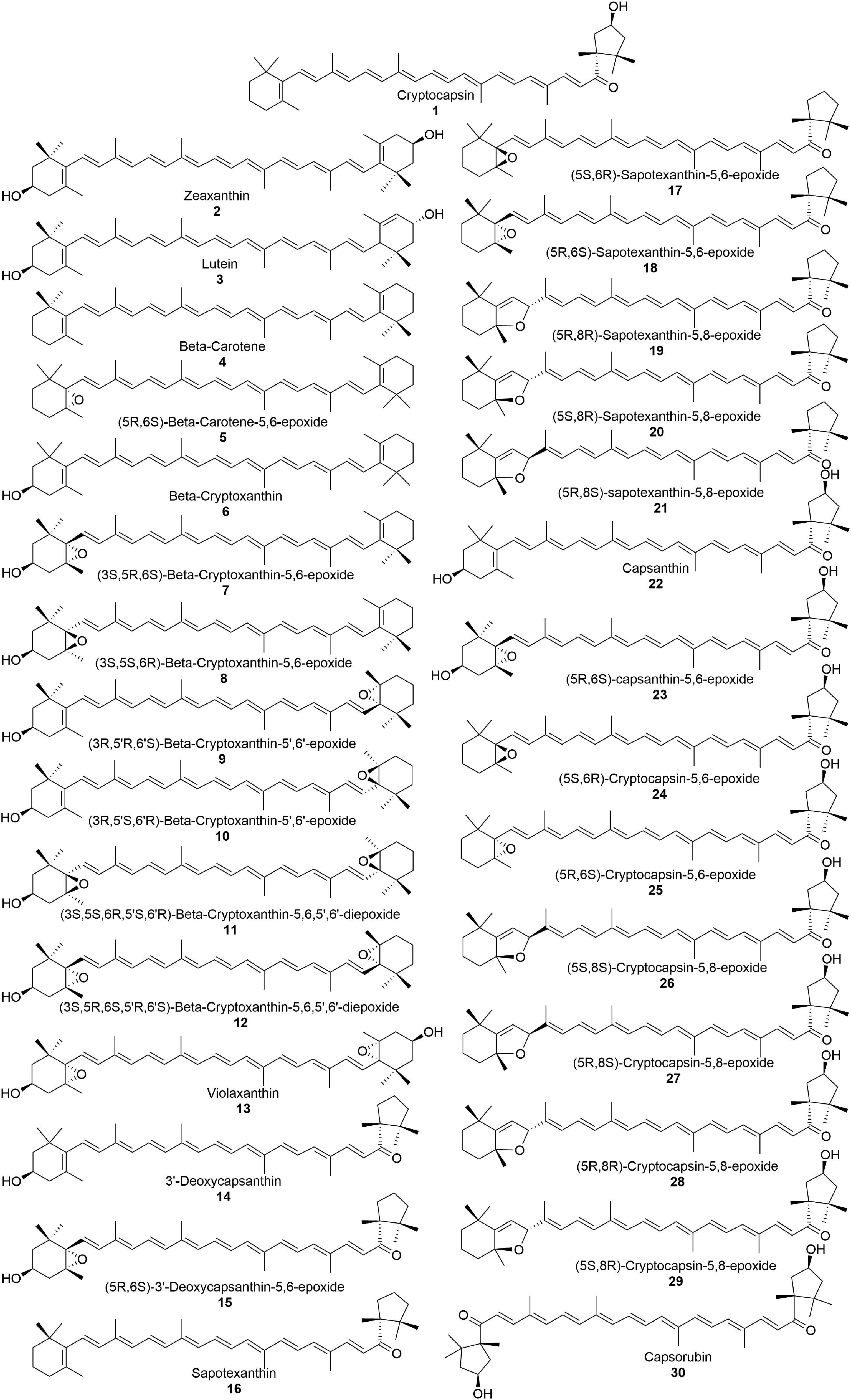
Carotenoids from P. sapota (red mamey fruit) selected for computational analysis.
We conducted molecular docking and molecular dynamic (MD) experiments on the interaction of thirty carotenoids from mamey fruit with Aβ40 and Aβ42 monomers to predict potential molecules that block Aβ aggregation kinetics. Using computational tools, we aimed to identify the most likely carotenoid(s) that can prevent the formation of Aβ aggregation. The findings from this study may guide the selective extraction of natural sources or facilitate semisynthetic carotenoids for future preclinical trials.
Methods
The aim of our study was to investigate the anti-aggregation properties of a series of carotenoids derived from natural products against the Aβ peptide. The first step involved performing geometry optimization and charge calculations for these carotenoids using density functional dispersion-corrected density functional theory (DFT) calculations with the B3LYP-D3 functional and 6–31 + g(d) basis set. To account for solvent effects, we employed the SMD model. 33 All calculations were conducted using Gaussian 16 software. 34
To account for the flexibility of the Aβ peptide, which is an important factor in studying anti-aggregation properties, we employed an MD protocol based on methodology described by Orjuela et al. 14 Three Aβ-peptide models were used: Aβ40 (PDB code: 1AML), 35 Aβ40 (PDB code: 1BA4), 36 and Aβ42 (PDB code: 1Z0Q). 37 Through exploring the conformational space of these models, we employed a robust molecular docking protocol, utilizing multiple computational packages to mitigate biases in the scoring function and ensure reliable results.
Molecular dynamics protocol of Aβ peptide
To capture the diverse range of conformations exhibited by the Aβ peptide and simulate the interactions with carotenoids over time, we performed MD simulations using the following parameters. The simulations were conducted using the Amber 18 packages. 38 The Aβ-peptide models were described using the FF14SB force field, 39 while the solvent system consisted of a periodic box of water with a NaCl concentration of 0.1 nM, employing the TIP3P force field. 40
The simulation protocol began with a 2 ns MD simulation in a canonical (NVT) ensemble to gradually heat the system from 0 to 300 K. Subsequently, the system density was equilibrated through a 5 ns MD simulation in an isothermal-isobaric ensemble. Finally, a 100 ns production phase was performed using an NVT ensemble to explore the conformational space of the Aβ peptide.
To identify representative conformations from the production phase, we employed the CPPTRAJ package 41 and applied the K-means algorithm 42 with 10 centroids, utilizing RMSD as the distance metric. All obtained conformations were analyzed using PyMOL 43 for further characterization and visualization.
Molecular docking of Aβ peptide
Considering all representative conformations of models of Aβ40/42 to molecular docking with carotenoids to calculate the carotenoid binding to every Aβ peptide model, we used a weighted scoring function for each carotenoid using the percentage of the individual representation with the following equation:

The choice of IZOQ and 1AML for the analyses was based on their specific structural characteristics relevant to our study. Despite their low percentile ranks in the PDB, these structures possess crucial features for understanding the interactions under investigation. Additionally, 100 ns of molecular dynamics simulations were performed to relax the structures at physiological pH, improving several PDB percentiles. Although other structures exist, these models have been previously used in similar studies. Therefore, IZOQ and 1AML were considered the most appropriate to ensure the reliability and relevance of our findings.
We considered that these 100 ns simulations were sufficient to achieve convergence and obtain reliable results consistent with experimental observations. Additionally, 10 clusters were obtained for each model to perform the molecular dynamics, ensuring a thorough sampling of the conformational space. Therefore, we determined that 100 ns was adequate for the current study. This approach balances computational efficiency with the accuracy needed to capture the relevant dynamics and interactions. In previous studies, the same models of Aβ peptides were used with 100 ns of molecular dynamics to reproduce the experimental studies on the anti-aggregating properties of carotenoids. 14
Prediction of physicochemical and ADMET properties
The SwissADME is widely used for predicting physicochemical properties and we used it to analyze 30 compounds. We also used an ADMET Predictor® in order to determine these properties.52,53 Lipinski's rules were used to analyze the compounds that do not respect the threshold values of these principles, including a molecular weight of less than 500 g/mol, no more than 5 donor bonds, no more than 10 acceptor bonds, and a partition coefficient (LogP) no more than 5 and routable bonds equal or minor of 5. If it complies, it will have adequate passive diffusion across cell membranes and effective absorption from the intestine to the blood. If more than two do not comply, poor absorption and permeability are expected. This rule is used for oral administration of drug.
In other way, Veber's rule states that a compound that has 10 or fewer rotatable bonds and a polar surface area no greater than 140 Å2 is likely to exhibit good oral bioavailability and less than 60 Å2 good permeability in brain membrane.
ADMET analysis
Software ADMET Predictor® version 10.4 (Simulations Plus Inc., Lancaster, CA, USA) was used as in silico simulation tool. All compounds were subjected to an ADMET analysis for the purpose of gaining insight into the biopharmaceutics and pharmacokinetic properties which includes prediction of several properties such as the absorption, distribution, metabolism, excretion, and toxicity.52–55 Simulations of nine selected carotenoids was performed with a human oral dose of 10 mg until 120 h. For each structure various pharmacokinetic parameters were assessed, namely, fraction absorbed (Fa%), bioavailability (F%), Human volume of distribution (Vd), and clearance (L/h) and ADMET parameters. These evaluations offer interesting details into the potential behavior and suitability of the compounds for further development.
Results
Aβ monomer molecular dynamics
To consider the high flexibility of the Aβ peptide, we started from a representative structure of the model reported in PDB (code: 1AML). This is a model of Aβ40 for which the MD simulations showed a significant fluctuation in three regions. The first region, from Asp1 to Tyr10, describes a nonstructural region. The second region, from Gly25 to Gly29, relates to a random coil. Finally, the third/last region, from Val35 to Val40, describes another nonstructural region, as shown in Supplemental Figure 1. In some representative structures, the α-helices were unfolded (Supplemental Figure 2). In comparison, the Aβ40 1BA4 and 1Z0Q models exhibited the same behavior in the first and last regions as the 1AML model. However, the random coil was less mobile in these models than the 1AML model. This difference is clearly seen in the positions of the α-helices within the model representations in Supplemental Figure 2.
Molecular docking
The results of the molecular docking simulations are presented in Table 1. The carotenoids (5R,8R)-sapotexanthin-5,8-epoxide (
Exponential consensus ranking (ECR) of carotenoids and Aβ peptide.
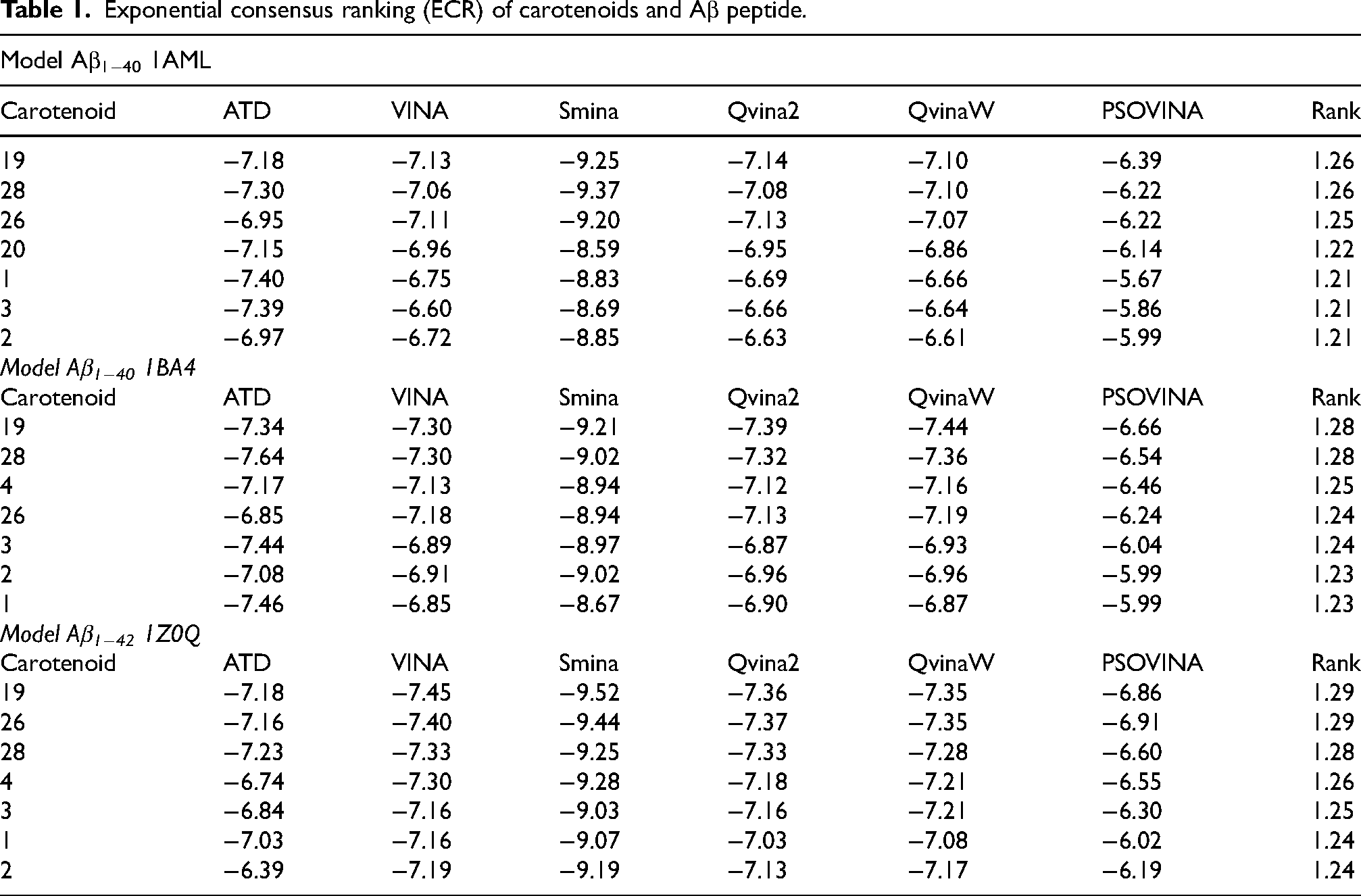
The molecular docking calculations performed for the Aβ42 peptide model (1Z0Q) exhibit a strong correlation with the experimental data of the carotenoids cryptocapsin (
In all models of the Aβ peptide, (5R,8R)-sapotexanthin-5,8-epoxide (
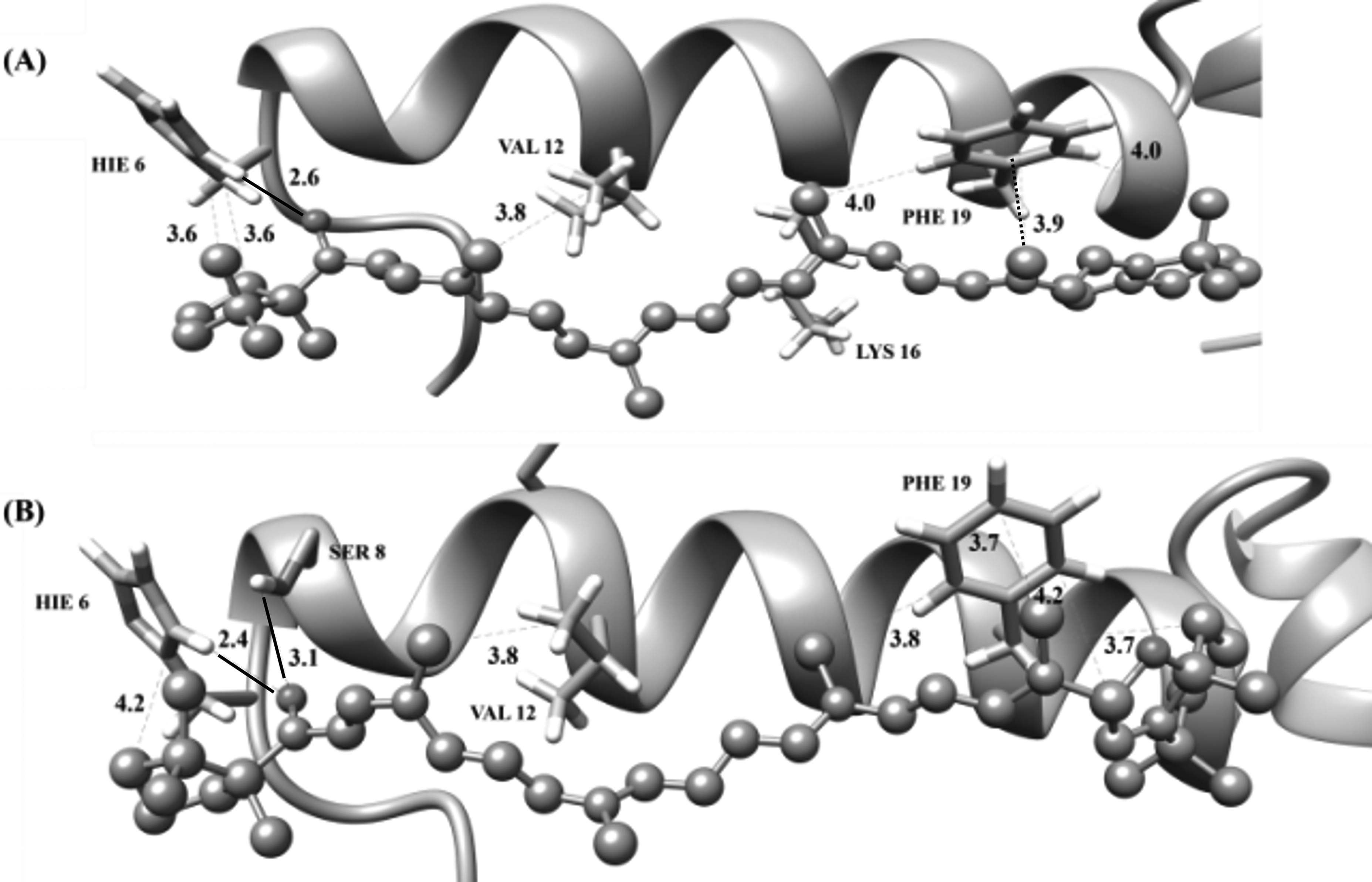
(a) Sapotexanthin (
When the interaction of carotenoid
Carotenoid
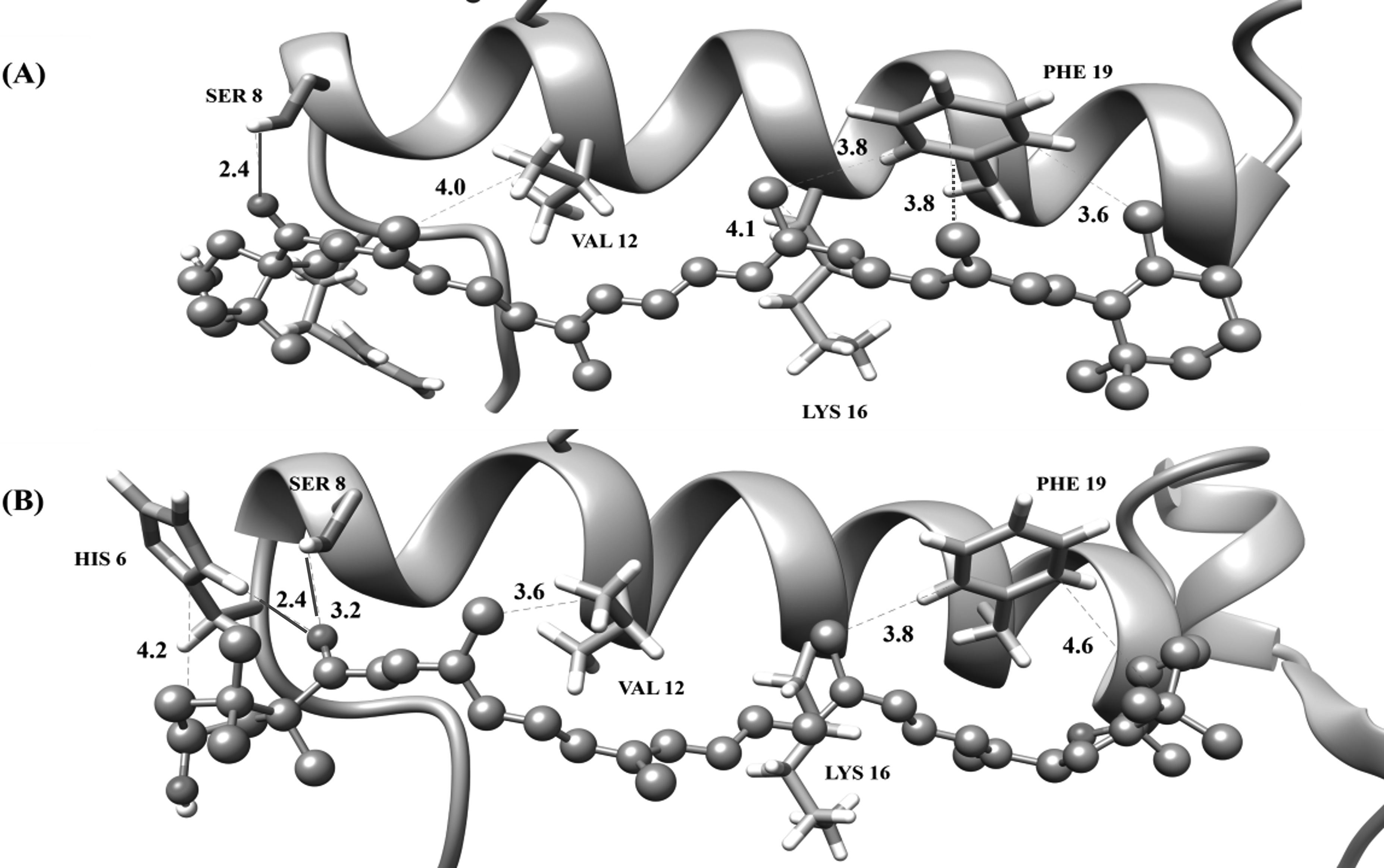
(a) Cryptocapsin (
Regarding carotenoid
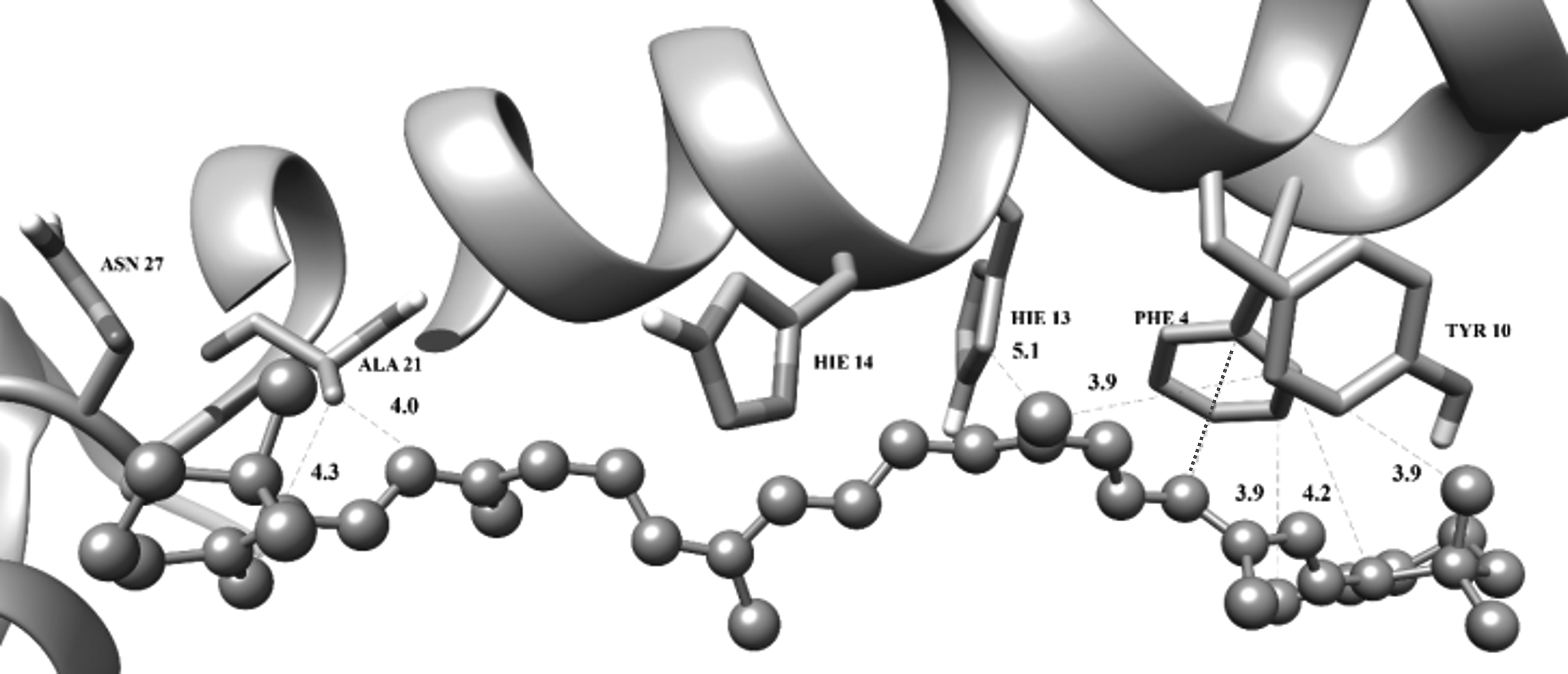
β-carotene (
For carotenoid

Lutein interaction with Aβ42: line (H-bond), dash line (alkyl-alkyl interaction). Distances are in angstroms.
Based on this computational analysis, we can observe three regions (a-c) important in cryptocapsin (Figure 7): region
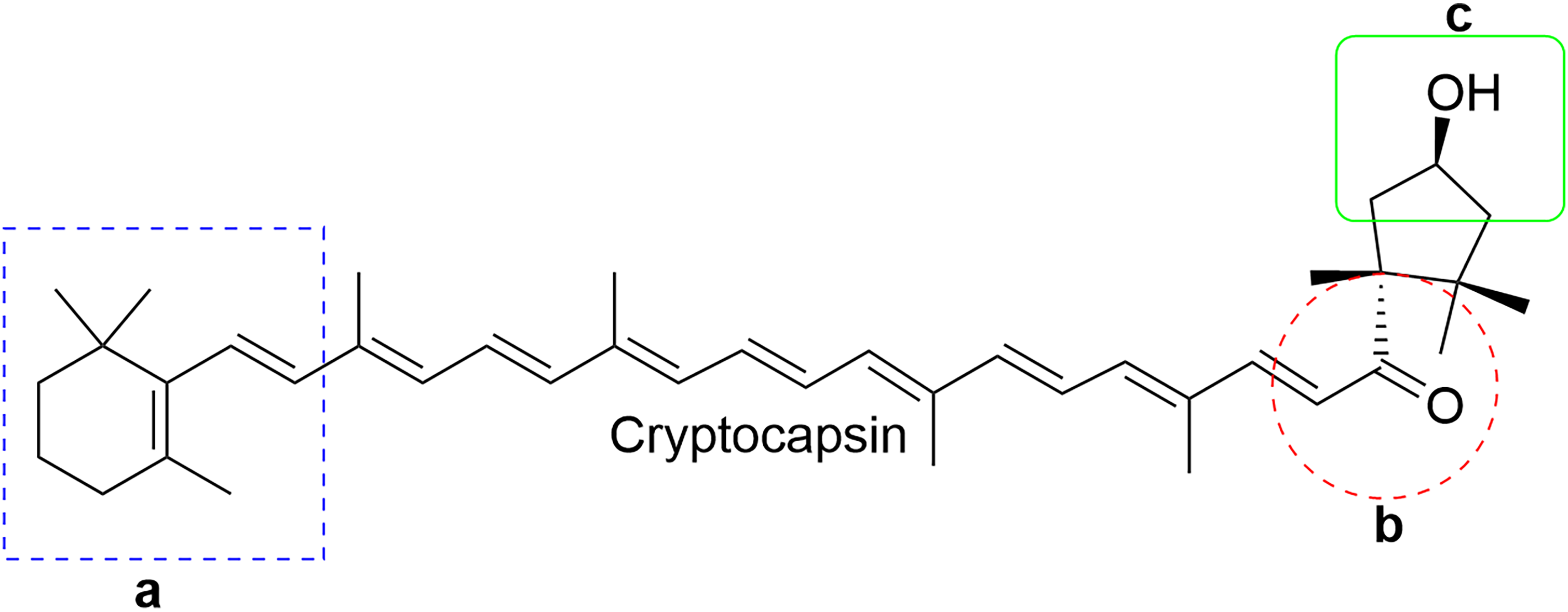
Cryptocapsin area essential for biological activity by computational analysis.
Prediction of physicochemical and ADMET properties
We predicted the physicochemical properties of 30 compounds. Results are shown in the Supplemental Material. The results of the physicochemical properties prediction of nine selected compounds, according to stronger biological activity, are shown in Table 2. For all nine compounds the water solubility was between 1.31 × 10−4 to 1.599 × 10−6, brain/blood partition coefficient (Log BB) were more than 0.3, molecular weight more than 500 g/mol, donor bonds no more than 5, acceptor bonds no more than 10, a partition coefficient (LogP) were between 8.2 to 11.6 and routable bonds more than 5. Zeaxanthin (
Prediction of the physicochemical properties of carotenoids analyzed using SwissADME® and ADMET Predictor®.

WS: Water solubility; Log BB: Brain blood partition coefficient; MW: Molecular weight; HBA: Hydrogen acceptor bonds; HBD: Hydrogen donor bonds; TPSA: Topological Polar Surface Area; RB: Rotatable bonds.
In the following table, the formula, water solubility, molecular weight, hydrogen acceptor bonds, hydrogen donor bonds and topological polar surface area report the same results in ADMET Predictor® and SwissADME. On the other hand, the Log P obtained differs slightly in both software's and the brain blood partition coefficient was obtained only with ADMET Predictor®.
Prediction of ADMET properties
We predicted physicochemical properties of 30 compounds. Results are shown in the Supplemental Material. Based on the Rank score, compounds cryptocapsin (
Prediction of carotenoid absorption and distribution processes analyzed using ADMET predictor® software.
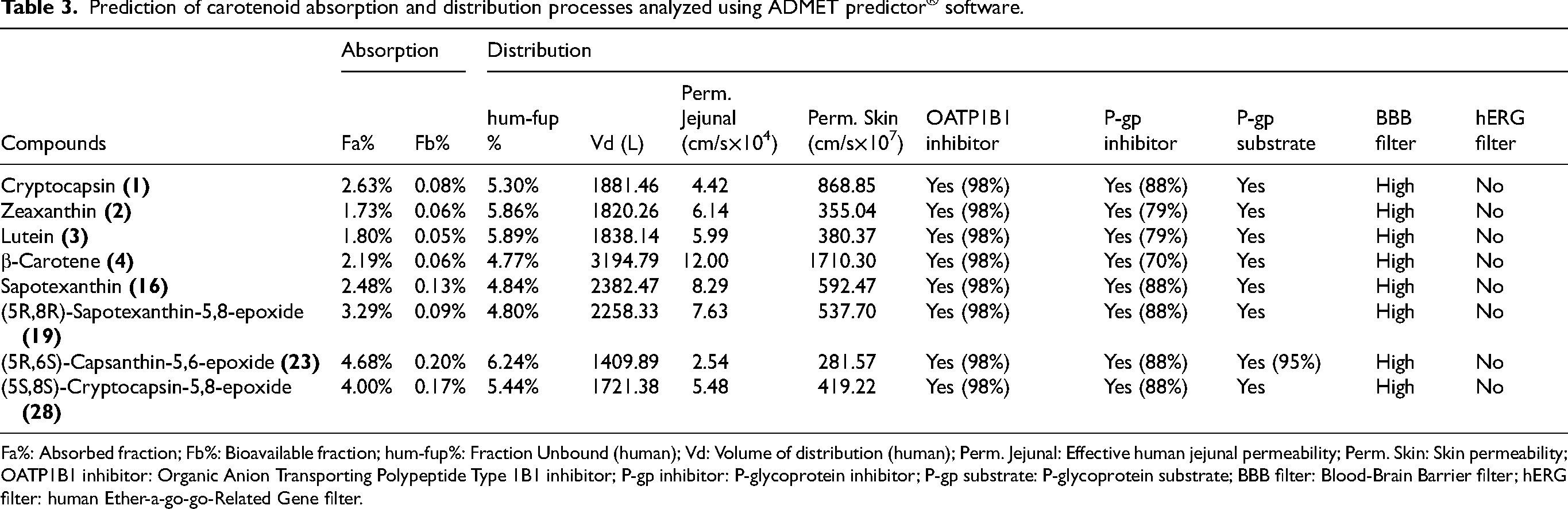
Fa%: Absorbed fraction; Fb%: Bioavailable fraction; hum-fup%: Fraction Unbound (human); Vd: Volume of distribution (human); Perm. Jejunal: Effective human jejunal permeability; Perm. Skin: Skin permeability; OATP1B1 inhibitor: Organic Anion Transporting Polypeptide Type 1B1 inhibitor; P-gp inhibitor: P-glycoprotein inhibitor; P-gp substrate: P-glycoprotein substrate; BBB filter: Blood-Brain Barrier filter; hERG filter: human Ether-a-go-go-Related Gene filter.
Prediction of carotenoid metabolism and excretion processes analyzed using ADMET Predictor® software.
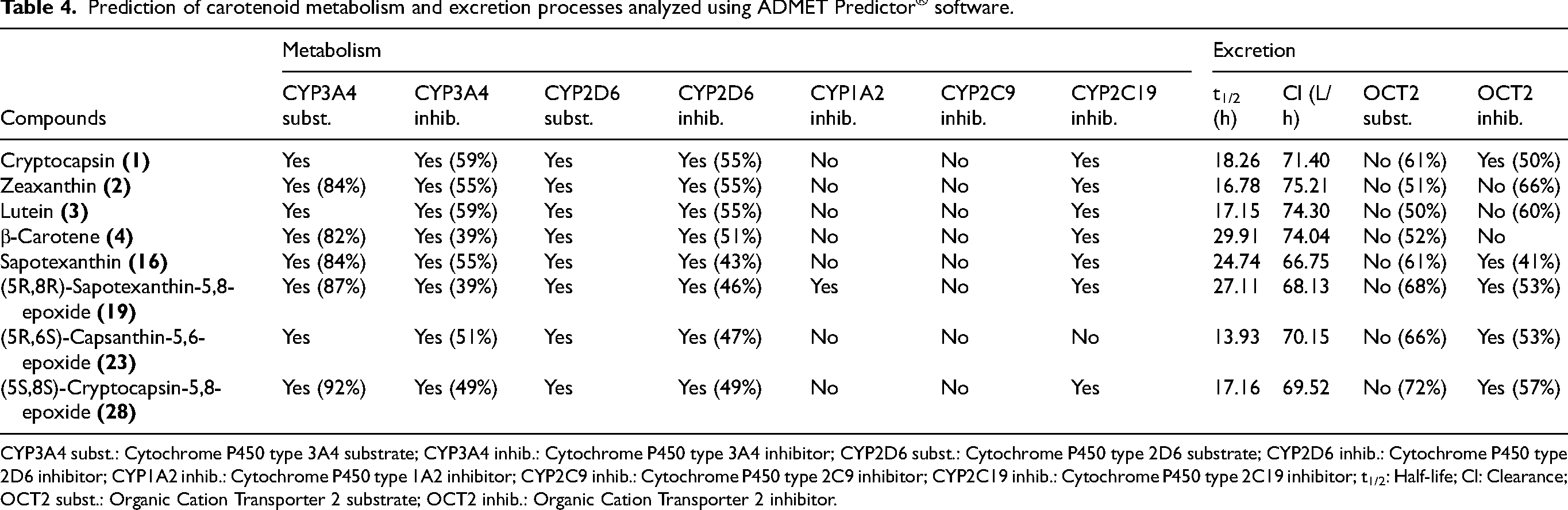
CYP3A4 subst.: Cytochrome P450 type 3A4 substrate; CYP3A4 inhib.: Cytochrome P450 type 3A4 inhibitor; CYP2D6 subst.: Cytochrome P450 type 2D6 substrate; CYP2D6 inhib.: Cytochrome P450 type 2D6 inhibitor; CYP1A2 inhib.: Cytochrome P450 type 1A2 inhibitor; CYP2C9 inhib.: Cytochrome P450 type 2C9 inhibitor; CYP2C19 inhib.: Cytochrome P450 type 2C19 inhibitor; t1/2: Half-life; Cl: Clearance; OCT2 subst.: Organic Cation Transporter 2 substrate; OCT2 inhib.: Organic Cation Transporter 2 inhibitor.
Prediction of the toxicity of carotenoids analyzed using ADMET predictor® software.
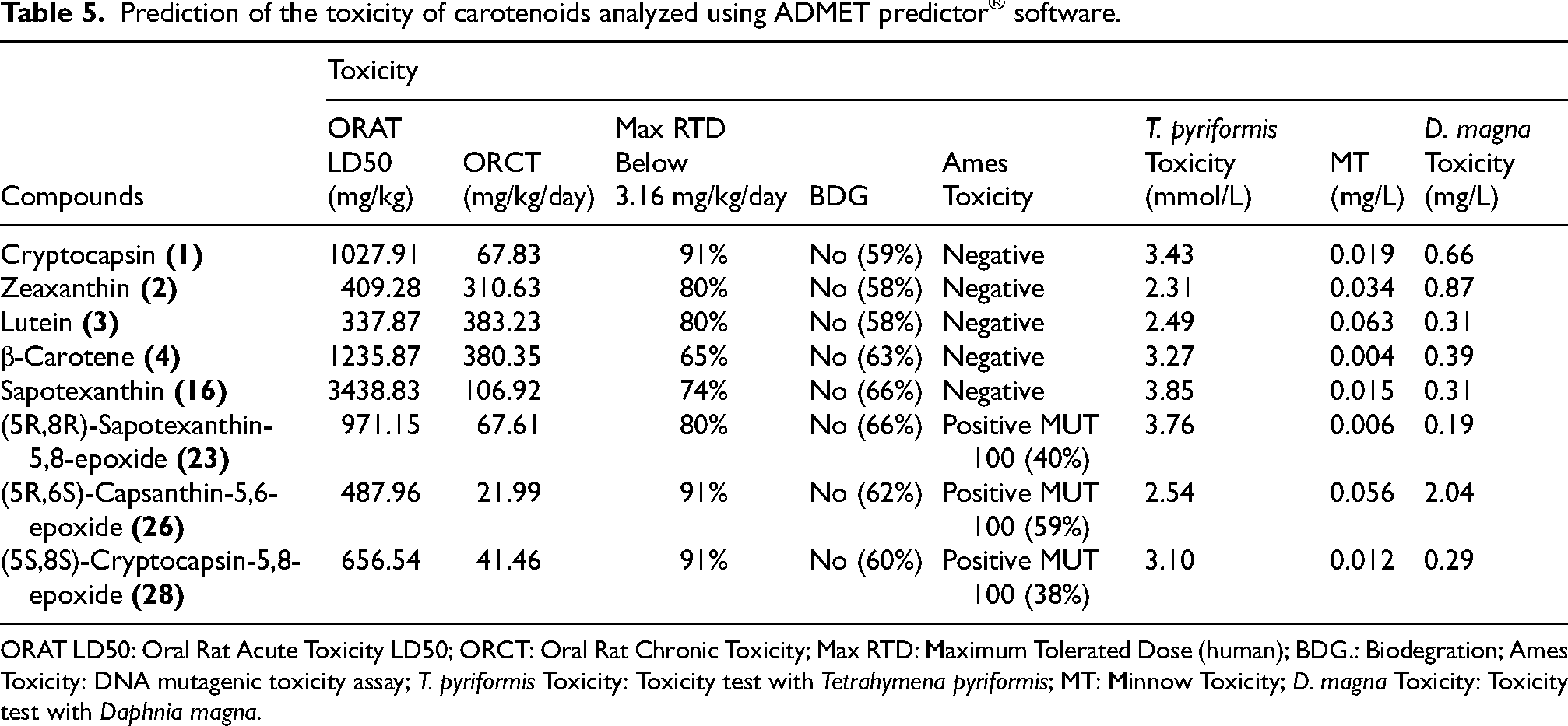
ORAT LD50: Oral Rat Acute Toxicity LD50; ORCT: Oral Rat Chronic Toxicity; Max RTD: Maximum Tolerated Dose (human); BDG.: Biodegration; Ames Toxicity: DNA mutagenic toxicity assay; T. pyriformis Toxicity: Toxicity test with Tetrahymena pyriformis; MT: Minnow Toxicity; D. magna Toxicity: Toxicity test with Daphnia magna.
Prediction of pharmacokinetic profiles after simulation of oral administration of all compounds (Figure 8) presented high volume of distribution and plasma concentration decrease gradually with the half-life of the compound longer. Compounds (5R,6S)-Capsanthin-5,6-epoxide (

Simulated human plasma concentration versus time after oral administration of 10 mg of each carotenoid.
The selected test compounds Cryptocapsin (
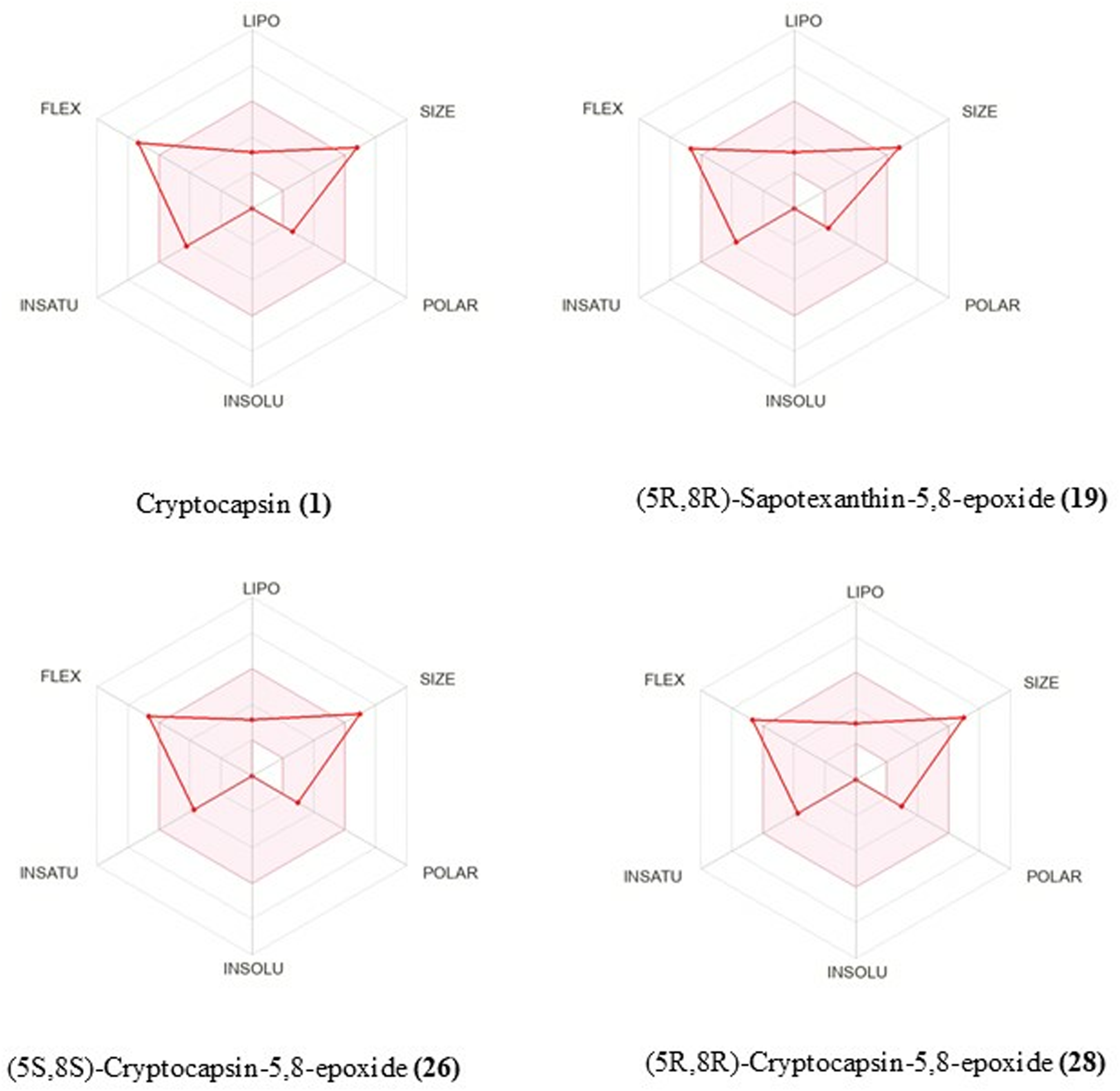
Radar plot of carotenoids selected.
Discussion
In the current computational study, we sought to clarify the interaction between Aβ peptides and conventional and unconventional carotenoids, focusing on their potential anti-aggregation properties. Our previous research showed that cryptocapsin, followed by zeaxanthin and cryptocapsin-5,6-epoxide, exhibited significant biological activity against Aβ aggregation.
19
Katayama et al. found that lutein was more biologically active than β-cryptoxanthin, β-carotene, α-carotene, and zeaxanthin.
22
Furthermore, our previous computational analysis
20
suggests that lutein was superior to cryptocapsin (
We observed a strong correlation between our computational results and the experimental data. Cryptocapsin (

Exponential consensus ranking (ECR) of model 1Z0Q Aβ42 numbered according to Figure 2. Systems 1, 2, and 3 are the experimental references.
The computational analysis revealed that the activity decreased in the following order: (5R,8R)-sapotexanthin-5,8-epoxide (

Comparison between base structures exponential consensus ranking (ECR).
Figure 12 shows a distinct variation in activity between the base carotenoid structures of conventional and unconventional carotenoids. Among the conventional carotenoids, β-carotene (
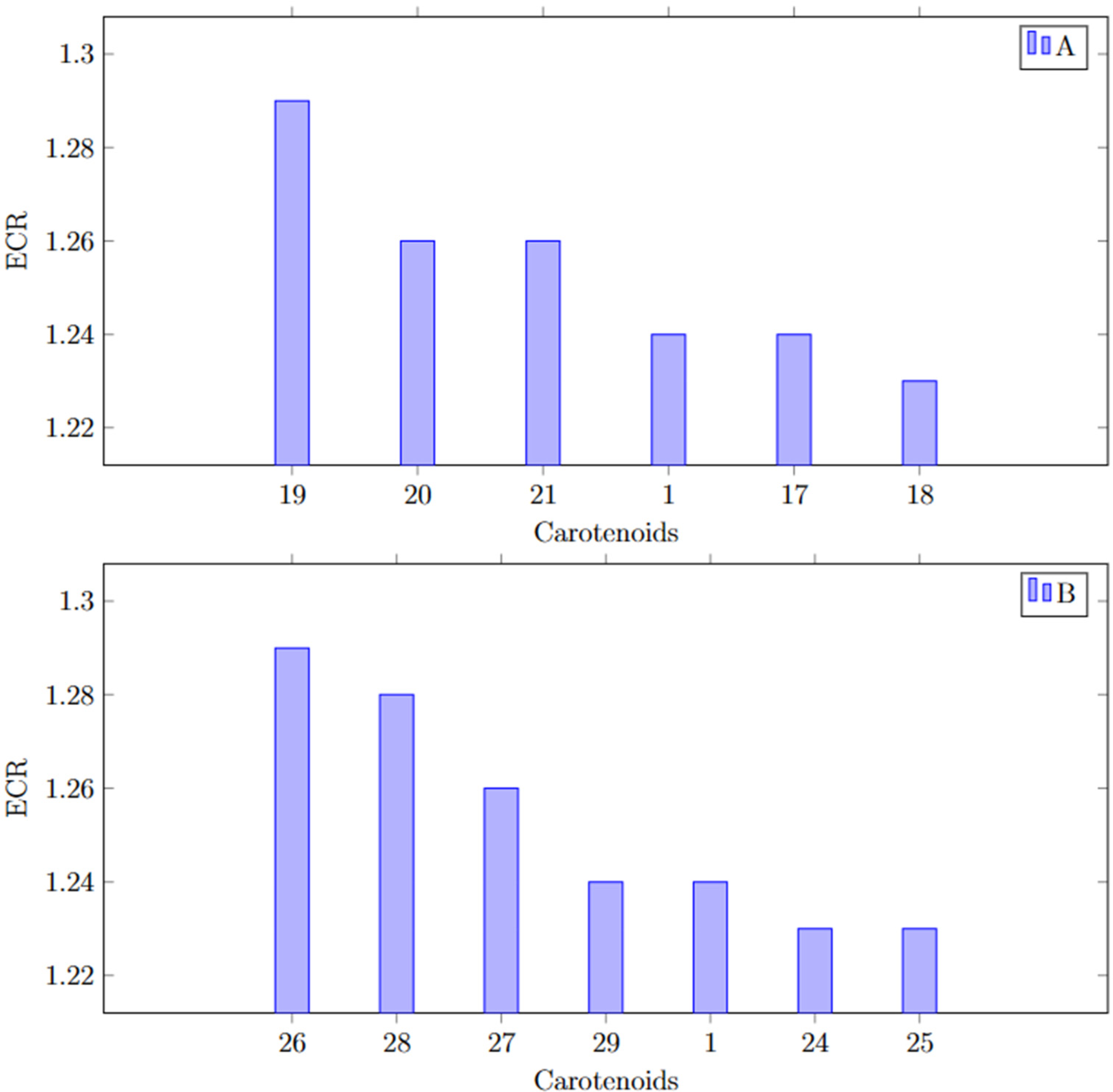
Exponential consensus ranking (ECR) of most active carotenoid derivates compared to cryptocapsin (
Notably, compared to the other 5,6 epoxidations, the 5,8 epoxidations of sapotexanthin and cryptocapsin exhibited greater promise Figure 12. Our results also suggested that the type of epoxidation could influence the anti-aggregation potential against Aβ42. Compared to the 5,6-epoxidation in sapotexanthin and cryptocapsin, 5,8-epoxidation appeared more promising, indicating the possibility of adjusting the activity via different epoxidation types. Moreover, stereochemistry was also influential; for instance, (5R,8R)-sapotexanthin-5,8-epoxide demonstrated greater activity than that of its (5S,8R) and (5S,8S) counterparts.
Regarding sapotexanthin-5,8-epoxide, variations in activity were observed among different forms as follows: (5R,8R) > (5S,8R) > (5S,8S). Similarly, cryptocapsin-5,8-epoxide exhibited distinct activity patterns as follows: (5S,8S) > (5R,8R) > (5R,8S). Notably, all forms of sapotexanthin-5,8-epoxide demonstrated higher activity than that of cryptocapsin, indicating the importance of conducting in vitro analyses to validate these in silico findings. Additionally, three variants of cryptocapsin-5,8-epoxide exhibited higher activity than cryptocapsin itself. Conversely, sapotexanthin-5,6-epoxide, cryptocapsin-5,6-epoxide, and β-carotene-5,6-epoxide displayed lower activity than cryptocapsin Figure 11, highlighting that the type of epoxide influences their respective activities. These molecular analyses will guide future in vitro investigations, which will focus on sapotexanthin-5,8-epoxide and cryptocapsin-5,8-epoxide.19,25,31,56–59 Computational analysis helps us prioritize carotenoids with higher potential therapeutic properties, thus saving considerable time and effort in the research process.
We also examined the possible chemical interactions between each carotenoid and the Aβ monomers. Our aim was to determine whether these selected carotenoids from red mamey fruit could block the self-assembly of Aβ aggregates. Our findings suggest that the biological activity and molecular docking of selected carotenoids are correlated with Aβ monomers. Computational analysis indicated that specific modifications to the terminal side of the carotenoid structure could significantly affect biological activity.
Based on that, the different in biological activity between Sapotexanthin (
Further studies are needed to experimentally validate these computational results. This study underscores the potential for harnessing natural compounds in the treatment of complex diseases. While synthesized compounds play a crucial role in modern pharmacology, nature also offers a vast array of complex molecules that may possess therapeutic properties. In this context, red mamey fruit and other rich sources of carotenoids could become vital resources in the development of new drugs to treat AD.
Physicochemical results indicate that these compounds do not adhere to the Lipinskís rules because more than two values are not complied, that means these compounds could have low absorption and permeability trough passive diffusion and suggest that mediated active transport is necessary. This is due to their low water solubility and their molecular weight of more than 500 g/mol. However, Veber's rule states that Cryptocapsin (
From biopharmaceutical point of view, the higher value of Log P indicates that more lipophilic is the compound and present high solubility in fat, oils, lipids and nonpolar solvents. Carotenoids showed high Log P that suggests compound could present limitations in passive diffusion and oral absorption. This suggest that mediated active transport is necessary for achieve absorption of the compounds by oral route. In the context of pharmacodynamics, the hydrophobic effect is the major driving force for the binding of drugs to their target receptors. On the other hand, hydrophobic drugs tend to be more toxic because they are generally retained for a longer time, have a wider distribution within the body (e.g., intracellular), are somewhat less selective in their binding to proteins, and finally are often extensively metabolized.
Inhibition of the three major cytochrome P450 (CYP) isoforms (CYP2C19, CYP2D6, and CYP3A4) could be certainly one major cause of pharmacokinetic-related drug–drug interactions and it were expected for our derivatives due to their physicochemical properties (large and lipophilic molecules). This point is important taking in account because drug-drug interactions (DDIs) and inhibition by one drug may lead to systemic toxicity due to elevated levels in the blood or subtherapeutic levels of another in the liver. Molecules were not CYP1A2 and CYP2C9 inhibitors, then there are selective inhibitions for CYP isoforms and administration of this molecules have to take care for specific drug and avoid together consumption. But also, is important recognize that process enzymatically converts lipid-soluble compounds to more water-soluble compounds to facilitate the excretion from the body.
For future in vivo studies it is important to evaluate other routes of administration as dermal route, since these derivatives showed good skin permeability, as well as new drug delivery systems in order to efficiency transport derivatives to achieve the desired action and avoid possible side effects. If oral route is preferred, then suitable drug formulations is required in order to assess the best oral absorption with the require deposition and activity.
Finally, this study provides an important advancement in our knowledge on the potential role of carotenoids in neurodegenerative disease. However, it is important to note that computational analysis is only the beginning. The promising candidates identified in this research must be validated through further in vitro and in vivo studies. If these subsequent studies confirm our findings, these compounds could become a valuable addition to the development of new treatments for AD.
Conclusions
According to computational and pharmaceutical analysis, compounds
Supplemental Material
sj-docx-1-alz-10.1177_13872877241291172 - Supplemental material for The role of carotenoids from red mamey fruit (Pouteria sapota) against amyloid-β monomers in Alzheimer's disease: Computational analysis and ADMET prediction
Supplemental material, sj-docx-1-alz-10.1177_13872877241291172 for The role of carotenoids from red mamey fruit (Pouteria sapota) against amyloid-β monomers in Alzheimer's disease: Computational analysis and ADMET prediction by Adrián L Orjuela, Marisín Pecchio, Jessica Cruz-Mora, Lakshmi Sowmya Emani, Maria B Carreira, Oleg V Larionov, Muralidhar L Hegde, K S Jagannatha Rao, Jorge Alí-Torres and Johant Lakey-Beitia in Journal of Alzheimer's Disease
Footnotes
Acknowledgments
M.P, M.B.C, J. A-T, and J.L.-B would like to acknowledge SENACYT Project [FID23-003]. K.S.J.R., O.V.L., and J.L.-B. would like to acknowledge SENACYT Project [APY-GC2018-010]. M.B.C and J.L.-B would like to acknowledge the National System of Investigation (SNI) for supporting their research. K.S.J.R. and J.L.-B. would like to acknowledge Melo Brain Grant (Panama) and INDICASAT Internal Grant [JR04-2020]. O.V.L. would like to acknowledge Welch Foundation grant number AX-0047. M.L.H is supported by the National Institute of Neurological Disorders and Stroke (NINDS) and the National Institute of Aging (NIA) of the National Institutes of Health (NIH) under award number RF1NS112719, and Everett E. and Randee K. Bernal for their support via Centenial Endowed Directorship of DNA Repair. A.L.O and J.A-T. want to thank Vicerrectoría de Investigación DIEB-UNAL, and the Center of Excellence in Scientific Computing (CoE–SciCo) for continuous support. LSE thankful to KELF for financial support through fellowship.
ORCID iDs
Author contributions
Adrián L Orjuela (Data curation; Formal analysis; Investigation; Methodology; Software; Validation; Writing – original draft; Writing – review & editing); Marisín Pecchio (Conceptualization; Formal analysis; Methodology; Software; Supervision; Visualization; Writing – original draft; Writing – review & editing); Jessica Cruz-Mora (Data curation; Formal analysis; Investigation; Methodology; Validation; Visualization; Writing – original draft); Lakshmi Sowmya Emani (Investigation; Visualization; Writing – original draft; Writing – review & editing); Maria B Carreira (Investigation; Visualization; Writing – original draft; Writing – review & editing); Oleg V Larionov (Supervision; Visualization; Writing – original draft; Writing – review & editing); Muralidhar L Hegde (Supervision; Visualization; Writing – original draft; Writing – review & editing); K S Jagannatha Rao (Conceptualization; Resources; Supervision; Writing – original draft; Writing – review & editing); Jorge Alí-Torres (Conceptualization; Formal analysis; Funding acquisition; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing); Johant Lakey-Beitia (Conceptualization; Formal analysis; Funding acquisition; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Secretaría Nacional de Ciencia, Tecnología e Innovación [SENACYT grant number [FID23-003], [APY-GC2018-010], Instituto de Investigaciones Científicas y Servicios de Alta Tecnología [INDICASAT AIP grant number [JR04-2020], the Welch Foundation grant number AX-0047, National Institute of Neurological Disorders and Stroke (NINDS), the National Institute of Aging (NIA) of the National Institutes of Health (NIH) under award number RF1NS112719, Everett E. and Randee K. Bernal for their support via Centenial Endowed Directorship of DNA Repair, Vicerrectoría de Investigación DIEB-UNAL, and the Center of Excellence in Scientific Computing (CoE–SciCo) for continuous support.
Declaration of conflicting interests
Jorge Ali-Torres is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
The other authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
All data generated or analyzed during this study are included in this published article and its supplemental material.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.