
Abstract
Background:
The most important hallmark in the neuropathology of Alzheimer’s disease (AD) is the formation of amyloid-β (Aβ) fibrils due to the misfolding/aggregation of the Aβ peptide. Preventing or reverting the aggregation process has been an active area of research. Naturally occurring products are a potential source of molecules that may be able to inhibit Aβ42 peptide aggregation. Recently, we and others reported the anti-aggregating properties of curcumin and some of its derivatives in vitro, presenting an important therapeutic avenue by enhancing these properties.
Objective:
To computationally assess the interaction between Aβ peptide and a set of curcumin derivatives previously explored in experimental assays.
Methods:
The interactions of ten ligands with Aβ monomers were studied by combining molecular dynamics and molecular docking simulations. We present the in silico evaluation of the interaction between these derivatives and the Aβ42 peptide, both in the monomeric and fibril forms.
Results:
The results show that a single substitution in curcumin could significantly enhance the interaction between the derivatives and the Aβ42 monomers when compared to a double substitution. In addition, the molecular docking simulations showed that the interaction between the curcumin derivatives and the Aβ42 monomers occur in a region critical for peptide aggregation.
Conclusion:
Results showed that a single substitution in curcumin improved the interaction of the ligands with the Aβ monomer more so than a double substitution. Our molecular docking studies thus provide important insights for further developing/validating novel curcumin-derived molecules with high therapeutic potential for AD.
Keywords
INTRODUCTION
Neurodegenerative diseases are characterized by structural modifications and misfolding of amyloidogenic proteins causing progressive neuronal death due to protein aggregation in the central nervous system [1–4]. The most common neurodegenerative diseases are Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS), [5–8]. AD is the most devastating neurodegenerative condition and a major health concern affecting more than 50 million people around the world [9, 10]. The characteristic features of AD are amyloid-β (Aβ) fibril accumulation in the brain, followed by tau protein phosphorylation and reactive oxygen species formation, among other neuronal processes, which lead to neuronal loss [11–16]. According to the amyloid hypothesis, Aβ fibrils are involved in triggering pathological consequences in AD [17, 18]. The aggregation pathway is characterized by the transformation of the Aβ monomer into an oligomer, which then forms proto-fibrils that finally lead to Aβ fibril formation[19–22]. AD is considered a complex and multifactorial disease caused by uncontrolled cleavage of the amyloid-β protein precursor (AβPP) by exogenous and endogenous factors that can play a role in triggering AD [11, 18]. Lifestyle factors, such as smoking, dietary deficiencies, and mental or physical inactivity may also contribute to the development of AD [16]. Furthermore, chronic co-morbid conditions, including diabetes, obesity, and cardiovascular diseases, are also linked to AD [16, 24]. Oxidative stress, metal ion dyshomeostasis, Aβ42 peptide aggregation, and intracellular tau protein phosphorylation are considered endogenous factors related to AD progression [16]. AβPP is a trans-membrane multi-domain protein with multiple cellular activities and is present in three main isoforms: AβPP695, AβPP751, and AβPP770 [11, 25]. The excision of AβPP can occur via two pathways. In the amyloidal pathway, β-secretase cleaves the AβPP at Met671 releasing a fragment of the secreted AβPP and a 99-amino acid C terminal residue fragment (CTFβ). This fragment is then cleaved by the enzyme γ-secretase at Val711 and Ala713, leading to the formation of AβPP intracellular domain (AICD) and Aβ40 and Aβ42 peptides; the Aβ42 peptide is considered to have higher toxicity and can precipitate in the brain due to its hydrophobicity [26–31]. In the non-amyloidal pathway, AβPP is cleaved by α-secretase at Lys 687, releasing a fragment of the soluble AβPPα and an 83-amino acid C terminal residue fragment (CTFα). This fragment is then cleaved by the enzyme γ-secretase, leading to the formation of AICD and Aβ17–42 (also known as protein 3), which does not accumulate in the brain [32]. For this reason, the inhibition of Aβ fibril formation is considered a therapeutic target in the fight against neurodegenerative disorders [33, 34].
We have focused on natural products as therapeutic neuroprotective molecules in the prevention or treatment of AD [35–37]. Several studies on polyphenols give new hope for neuroprotective activity against AD [38, 39]. One such popular molecule is curcumin, a natural product with powerful therapeutic properties and the potential to form new alternative therapies against AD.
Curcumin is found in the rhizome of Curcuma longa and considered a pleotropic molecule. However, curcumin (Fig. 1) has low bioavailability due to its low solubility in water and its instability at physiological pH at which the molecule is transformed into ferrulic acid, vanillin, dehydrozingerone, and curcumin glucuronide. These factors present challenges in drug discovery with curcumin [40–42]. Despite these challenges, curcumin has demonstrated multiple therapeutic properties; our group has fo-cused on anti-aggregation and anti-inflammation properties in our efforts to explore alternative thera-peutic avenues for AD [35, 43]. Our recent publication focused on the synthesis of curcumin derivatives with the goal of enhancing bioavailability and main-taining neuroprotective activities [35]. We determined the anti-inflammatory properties of curcumin derivatives and established a structure-activity relationship [35]. Curcumin and its derivatives have demonstrated interesting results that can be improved if we can determine which chemical interactions are crucial for anti-aggregation activity. In our current study, we present molecular docking and molecular dynamic (MD) studies on the interaction of nine novel curcumin derivatives synthesized in our laboratory with Aβ40 and Aβ42 monomers and Aβ42 fibrils to identify molecules that can potentially block Aβ aggregation.
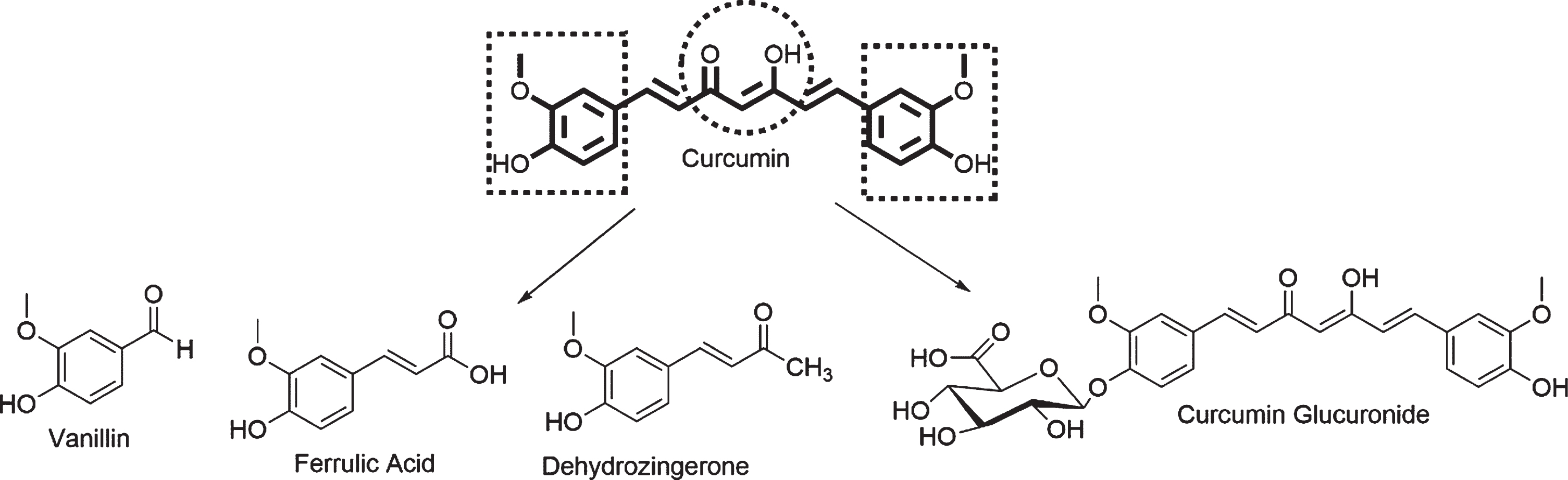
Curcumin and its metabolic products.
MATERIALS AND METHODS
Computational details
To study the interaction between curcumin derivatives (Fig. 2) and the Aβ40 and Aβ42 monomers and Aβ42 fibril models, we started with the novel curcumin derivatives reported previously by Lakey-Beitia et al. [35]. The geometries of these curcumin derivatives were optimized with the density functional dispersion corrected B3LYP-D3 and the 6–31 + g(d) basis set. In this optimization, solvent effect (water) was included as a continuum through the solvation based model [44]. All these calculations were performed in the Gaussian 16 program suite [45].

Curcumin derivatives used in this study.
To understand the molecular basis of the curcumin derivatives’ anti-aggregation properties, we carried out molecular docking simulations between ligands and three nuclear magnetic resonance (NMR) models of the Aβ peptide obtained from the Protein Data Bank (PDB): two Aβ40 models (PDB codes: 1AML, 1BA4) [46, 47] and one Aβ42 model (PDB code: 1Z0Q) [48] . Before completing the docking process, we conducted MD simulations with the three Aβ peptides to explore the most stable conformations and to account for the high flexibility in these structures.
In addition, to understand the interaction of these compounds with the Aβ42 fibrils, we evaluated their interaction with an Aβ42 fibril model. The details of these calculations are presented in the following sections.
Molecular dynamics simulations of Aβ monomers
Due to the high flexibility of Aβ monomers, we performed MD simulations to consider the most stable conformations in the docking process (ensemble docking). All MD simulations were carried out with the Amber 18 package [49]. The system was prepared by using tleap with the following parameters: Aβ monomers were described with the amber FF14SB force field [50], solvent was added with the TIP3P water model in a periodic box, and a sodium chloride (NaCl) concentration of 0.1 mM was considered. For each of the monomer, we ran 5 ns of MD to equilibrate the system and 5 replicas of 100 ns of production MD. The full molecular dynamics protocol is described in the Supplementary Material. To visualize the trajectories, we used Visual Molecular Dynamics (VMD) [51] and to analyze these trajectories, we used the CPPTRAJ package [52]. To select the most stable conformations for the molecular docking, we used the K-means cluster algorithm [53], using 10 centroids, and analyzed them with PyMOL [54].
Molecular docking between curcumin derivatives, Aβ monomers, and fibrils
Curcumin and its derivatives were docked against ten representative structures of every Aβ monomer model. The binding scores are presented as a weighted average from the ten conformations, determined by a k-mean algorithm using the Root Mean Square Distance (RMSD) similarity in all MD and an exponential consensus ranking (ECR) using three molecular docking programs: AutoDock 4 (AD4), AutoDock Vina (Vina), and Smina [55–57]. To explore the best binding site in the Aβ models, we used a consensus of three docking softwares: AD4 [58], Vina [56], and Smina (vinardo scoring function) [57]. The models were prepared in AutoDock Tools [58] to add Gasteiger charges and prepare the pdbqt format. The charges of the curcumin derivatives were taken from the Mulliken population analysis based on density functional theory calculations. The search space in the molecular docking simulations was selected around the full peptide of the Aβ monomer to determine the ideal position of the ligands. The interactions were analyzed with Maestro software [59]. Finally, to improve the docking outcomes, reducing the structure and system dependence [60], we used the ECR described by Palacio-Rodríguez et al. [61] by using the same three molecular docking programs. The ECR of each molecule was described using a consensus score P(i) corresponding to the sum of the exponential rank of j programs, following the equation below:
where σ is the expected value of the exponential distribution with respect to the number of molecules i, and
To observe the stability and conformational changes of the Aβ-curcumin derivate complex, we selected the best Aβ-derivate complex from the docking analysis; next, we used the same protocol of Aβ peptide MD, and the curcumin and derivates were described with a force field applying a quantum mechanical calculations parameter; the stability was studied using RMSD changes for all possible conformations of Aβ, the curcumin derivatives, and the Aβ+-curcurmin derivative.
To study the interactions between Aβ42 fibrils and curcumin derivatives, an NMR fibril model reported in the PDB (2BEG) [62] was used. We considered the first conformation reported in the PDB for the docking calculations, since the model does not show high flexibility. The model was protonated at pH 7 to simulate a physiological environment. Due to the rigidity of the Aβ42 fibril compared to the Aβ monomers, no MD was performed in order to conserve the structure extracted from the experimental report. Similar to the protocol we used with the Aβ monomers, to improve the accuracy of the energy and the ligand conformations in the molecular docking studies, we used a previously described rank consensus protocol [61] with same three molecular docking programs: AD4, Vina, and Smina. The charges of the ligands and fibril model were added using the same methodology described for the Aβ monomers. For the molecular docking simulations, we considered the entire Aβ fibril in our search for the best ligand positions.
RESULTS
This section is presented according to the system’s size. The results of the docking and MD simulations between the curcumin derivatives and the Aβ monomers are presented first, followed by the interaction between the derivatives and the fibrils model.
Conformational ensembles of the Aβ monomers
MD simulation of the Aβ monomers showed important changes in the Aβ peptide structure. The peptide’s initial alpha-helix structure was transformed into a structure with two alpha-helices and one random coil. Figure 3 shows the conformational changes of Aβ40 (1AML) and the percentage of time the monomer is present in each conformation; the representative structures for the 1BA4 and 1Z0Q models are shown in the Supplementary Material. As can be seen, in the representative structures 1, 2, 3, 6, 8, and 10, two alpha helix regions are observed: the first one from Glu11 to Asp23 and the second region from Ala30 to Val36. These two regions are linked by a random coil from Gly25 to Gly29. These amino acids are the same observed in the turn regions of the Aβ42 fibrils’ crystal structures (2BEG and 2MLN) reported in the PDB. This finding suggests that blocking this folding region could serve as a possible mechanism for aggregation inhibition.

Representative structures of the production phase of molecular dynamics for Aβ40 model 1AML.
Molecular docking between curcumin derivatives and Aβ monomers
Docking calculations often show that docking into un-complexed receptors produces poor results. Therefore, sometimes it is necessary to modify the position of the receptor side chains to increase the probability of successful docking calculations. Using MD or other sampling methods to generate different rotamers of the side chain, based on representative structures, have shown good results in docking and virtual screening campaigns [63–66]. Thus, by combining several starting structures and molecular dynamics, we were able to increase the probability of finding acceptable to Aβ40/42-curcumin derivative complexes. Therefore, molecular docking was performed on the ten most frequent structures extracted from the MD simulations for the models 1AML, 1BA4, and 1Z0Q.
The results of this docking process are presented in Table 1. The binding scores of the curcumin monosubstituted derivatives (ligands 2–5) were more favorable than those of the disubstituted derivatives for the three docking programs used. This is due to the presence of an H-bond established by the OH group from curcumin (Fig. 4). This interaction is not present in the disubstituted ligands. Additionally, as shown in Fig. 4, the ligands form dispersive-like interactions with side-chains of the non-polar amino acids present in the Aβ40 monomer. These interactions are key to preventing the formation of the β-sheet-like structure characteristics of the Aβ fibrils.
Weighted average score for molecular dynamics conformations of Aβ monomers and curcumin derivatives
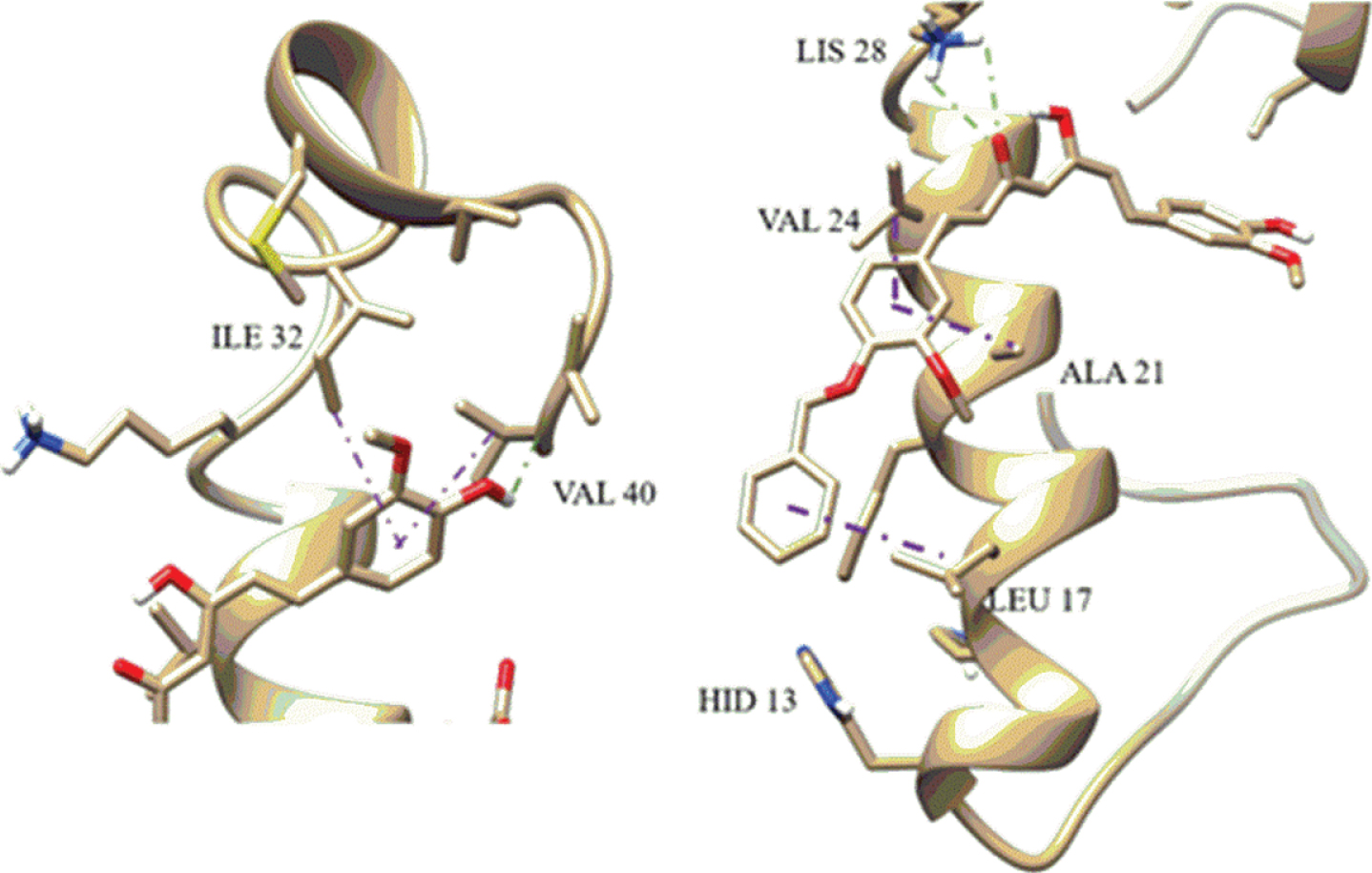
Example interaction between monosubstituted curcumin and Aβ40 peptide. Aβ-alkyl interaction is presented in purple and hydrogen-bond in green.
The docking outcomes are program and structure dependent. Thus, while docking software can show positive results for a specific structure, the same software can yield negative results for other receptor structures. To overcome this limitation, we implemented the exponential consensus rank (ECR) strategy using several docking software programs. Table 2 reports the ECR and the experimental activity of active compounds; in general, the ECR strategy shows a high degree of concordance with the IC50 values. As can be seen from this table, the monosubstituted ligands have better ranked interactions with the three Aβ40 monomers when compared to the disubstituted ligands. As mentioned before, these differences are due to the ability of the curcumin’s hydroxyl group to establish an H-bond interaction with the peptide. Additionally, the size of the derivatives increases with a second substitution, which generates steric hindrance to establishing a stabilizing interaction with the partially folded peptides; this hindrance reduces the number of contacts and hence, the interaction’s score value. An example of the interaction between one disubstituted curcumin derivative and the Aβ monomer is presented in Fig. 5.
Exponential consensus ranking (ECR) of curcumin derivatives and NMR models of Aβ monomers
N/A, not active.; N/P, not position.
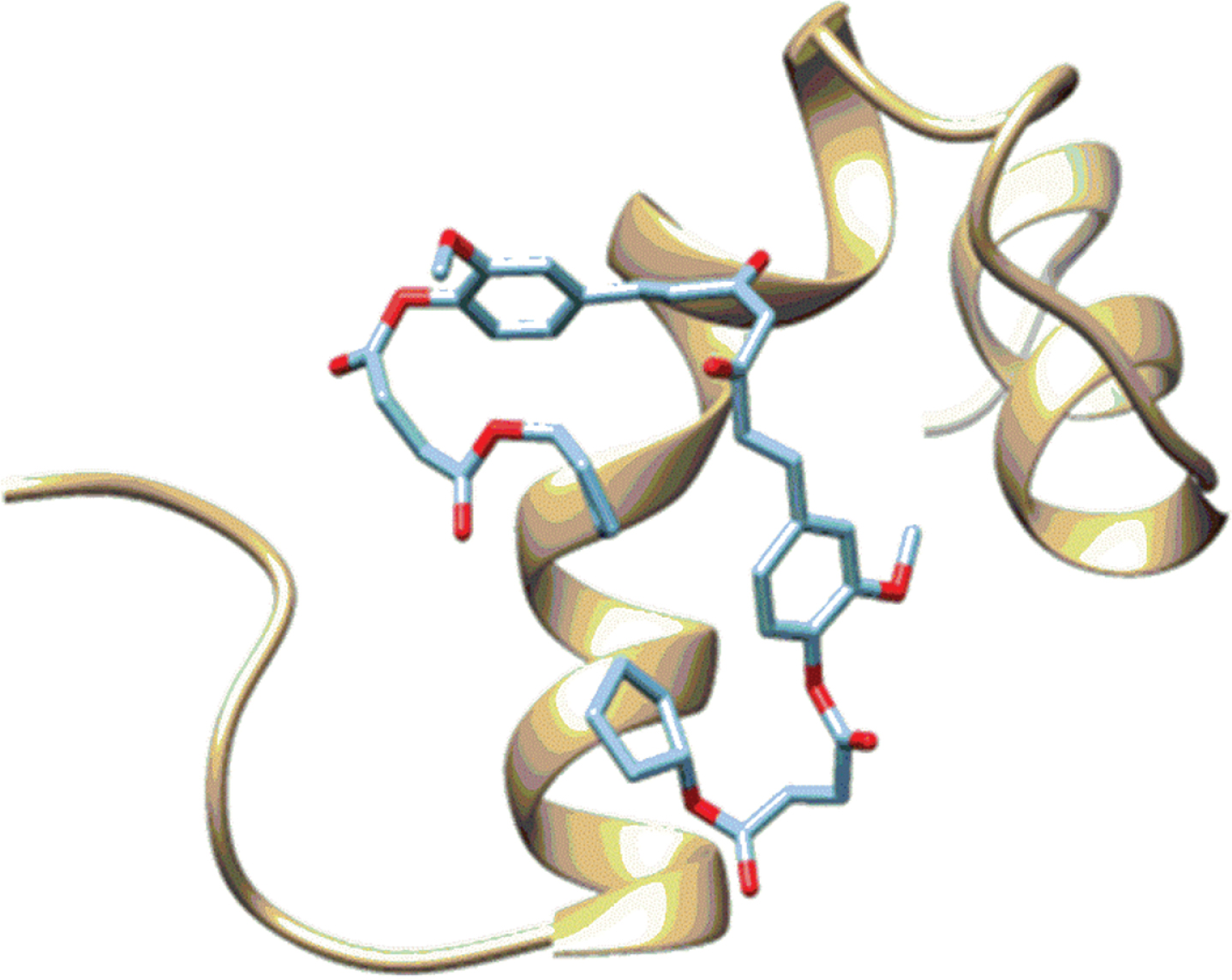
Example binding pose of disubstituted curcumin derivate (Cur(OSuccAllyl)2) with an Aβ model.
As can be seen from this figure, the ability to block the folding region further confirms the ability of these derivatives to prevent Aβ aggregation as has been observed in in vitro experiments. However, the disubstituted ligands have lower interactions with the Aβ monomer than the monosubstituted ones. The results of these simulations suggest that a single substitution in the curcumin molecule is sufficient to improve its activity.
Molecular dynamics between curcumin derivatives and Aβ40
In the MD, we used the highest ranked curcumin derivative CurOBn to show the derivative’s stability and interactions over time; in addition, we used the disubstituted curcumin derivative Cur(OSuccAllyl)2 and an unsubstituted curcumin derivative as a reference.
The RMSD along the five MD replicas are shown in Fig. 6. As can be seen from this figure, in all three cases, the Aβ40 is very flexible and fluctuated throughout the simulation. However, several interactions between the ligand and the peptide were maintained during the MD, demonstrating the stability of the peptide-ligand complex (Fig. 7). According to the MD of the three complexes, the complex containing the monosubstituted derivative shows a higher stabilization of the peptide than the unsubstituted curcumin complex and the disubstituted complex, as can be seen from the RMSD values.
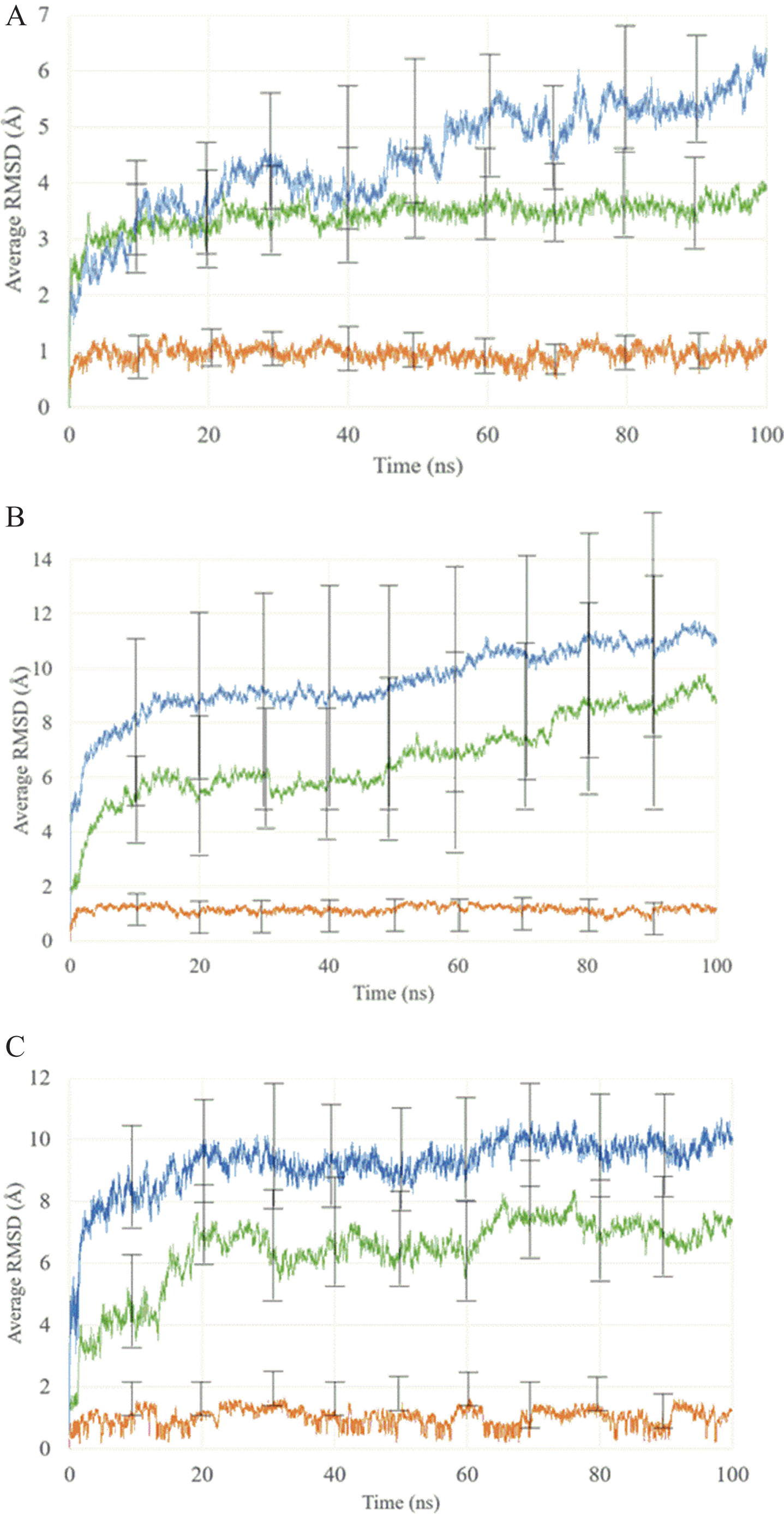
Average RMSD and error bars of 5 replicas MD of curcumin derivatives and Aβ40 peptide. In the graphics curcumin or its derivates are shown in orange, Aβ40 in green and complex Aβ-curcumin or derivative in blue. Graphic A is for CurOBn derivative, B for Cur(OSuccAllyl)2, and C for curcumin which is used as reference.

Structural changes of Aβ42 during the simulation time. Light blue initial structure obtained from docking simulations, yellow 25 ns, green 50 ns, and pink 75 ns of MD. A) Complex Aβ42 with Curcumin; B) Complex Aβ42 with CurOBn.
From Fig. 6 we observe that the ligands were stable over time with a change in the RMSD below 2Å. The peptide-ligand complex in all cases was stabilized in 20 ns of MD. The Aβ40-ligand complexes and Aβ40 exhibit the same dynamic behavior in all cases, but the Cur(OSuccAllyl)2 complex had structural changes close to 50 ns, 70 ns, and 80 ns as shown in Fig. 8. Considering the flexibility of Aβ40 demonstrated by our simulations, structural changes in the random coil and amino acid region 1–14 could affect the RMSD results. In our RMSD analysis for curcumin and CurOBn, the changes were constant across time; however, these changes were less significant for the curcumin derivatives with a stability structure shown in Fig. 9.

Structural changes in the molecular dynamics of Aβ40 1BA4 with Cur(OSuccAllyl)2.

Structural changes in the molecular dynamics of Aβ40 1BA4 with CurOBn.
Molecular docking between curcumin derivatives and Aβ42 fibrils
Molecular docking was performed between the curcumin derivatives and Aβ42 fibrils (PDB code: 2BEG) with Vina, AD4, and Smina. The same ECR protocol used for the Aβ monomers was implemented for this analysis. The results of the ECR are presented in Table 3 and Fig. 10. The ECR rank predicts that the monosubstituted ligands CurOBn, CurOSuccCyp, and CurOAc have the highest affinities toward the Aβ42 fibril. However, compounds with anti-aggregation activities (Curcumin, CurOSuccAllyl, and Cur(OSuccAllyl)2) were among the lowest ranked compounds, together with several non-active disubstituted curcumin derivatives. For instance, contrary to the results for the monomeric state, curcumin showed a lower ranking than a disubstituted derivative, Cur(OSuccBn)2, and did not demonstrate anti-aggregation activity.
Exponential consensus ranking (ECR) of curcumin derivatives and the Aβ42 fibrils
N/A, not active.; N/P, not position.

Fibril interaction with Aβ fibril 2BEG sing different programs. Left AutoDock Vina. Center Smina, Right AutoDock 4.
According to the IC50 and the molecular docking results, the ECR or the relative activities of the curcumin and its derivatives are better predicted by the docking performed with the Aβ monomers than the docking performed with the Aβ42 fibril. Thus, we hypothesize that the active compound shows more affinity for the Aβ monomers. Although these results can be influenced by the structure dependence of the docking programs, we reduced that dependence by implementing a consensus rank strategy. Thus, we have evidence to hypothesize that active curcumin compounds have a higher affinity for the Aβ monomers. Therefore, the monosubstituted curcumin derivative could stabilize the Aβ monomer, displacing the ECR Aβ42 fibril/Aβ monomers equilibrium toward the monomeric state, supporting the derivative’s anti-aggregation properties. However, the previous hypothesis must be tested with in vitro experiments or exhaustive simulations of the complexes, which is beyond the scope of this paper.
DISCUSSION
In this investigation, we were focused on finding a relationship between molecular docking and biological activity of curcumin derivatives with Aβ monomers and fibrils. We consider molecular docking a powerful tool that allows for rapid determination of the chemical interactions that are crucial in the aggregation process. With these results, we can design and synthesize novel compounds with therapeutic potential for neurodegenerative diseases. In our previous report, we showed the results of in vitro experiments indicating the anti-Aβ aggregation activity of curcumin and its derivatives [35]. When we applied molecular docking to the selected compounds, we observed the different chemical interactions that curcumin derivatives can have with Aβ monomers and/or fibrils. The simulations indicate that curcumin derivatives can block the self-assembly of Aβ monomers or facilitate the disaggregation of Aβ fibrils. According to the literature, amino acid residues in the Aβ16–20 region of Aβ42 have been shown to be relevant for the self-association of Aβ42 peptides into Aβ42 fibrils [2, 67]. In the Aβ fibril structure, Aβ16–20 forms an anti-parallel-sheet structure through binding to the homologous regions, Aβ17–21 or Aβ18–22. It has been shown that Aβ16–20 deletion or the binding of ligands to the Aβ16–20 region of Aβ42 inhibits the formation of Aβ42 fibrils [68, 69], which indicates that this region (16–20) is the most important nucleation site for Aβ42 aggregation.
Our results indicate that curcumin derivatives could inhibit aggregation by means of strong interactions with the 20–28 regions of Aβ peptides (Fig. 4). However, the curcumin derivatives show additional interactions that may disrupt aggregation; for example, hydrogen bonding at the salt bridge Lys28 disturbs the stability of the salt bridge between Aβ monomers (Fig. 4). Similarly, the derivatives also show interaction with the C terminal residues Ile32 and Val40 (Fig. 4). We observed similar behavior between docking model results with biological activity. We conclude that the curcumin derivatives with a hydroxyl group that is available to form H-bond interactions with Aβ monomers/fibrils have a higher potential to inhibit Aβ aggregation. Figure 2 shows the curcumin derivatives used in this investigation. Monosubstituted curcumin derivatives, such as CurOBn, CurOAc, CurOSuccAllyl, CurOSuccBn, and CurOSuccCyp, have a hydroxyl group to form an H-bond with Aβ. However, differences in the behavior of these derivatives indicate that it is also important to take into account the length of the molecules; the curcumin derivatives CurOSuccAllyl, CurOSuccBn and CurOSuccCyp are longer than CurOBn and CurOAc and show higher biological activity and greater H bonding interactions with Aβ. When the curcumin derivative is disubstituted, the biological activity is reduced until it is inactive. This inactivity was also observed in the molecular docking simulations where H-bond and π-π interactions with Aβ were not noted. With this result, we know that it is important to have monosubstituted instead disubstituted curcumin derivatives. Moreover, the monosubstituted derivatives should not have a connector because increasing the size of the molecule reduces its activity. This result was also evident in the anti-inflammatory bioassay. This indicates the importance of the curcumin derivative’s size, for instance, short derivatives with one hydroxyl group have the potential to increase the π-π interaction with Aβ. We found agreement between our molecular docking results and our experimental results. This relationship will help us to design novel molecules with neuroprotective activity.
Conclusions
Molecular docking is a valuable tool in identifying a novel anti-amyloidogenic molecule/structural derivative with therapeutic properties against AD. A combination of MD and molecular docking simulations were used to evaluate curcumin and its derivatives for their interaction with Aβ. Results showed that a single substitution on curcumin is better than a double substitution to improve the interaction of the ligands with the Aβ monomer. A relationship between molecular docking and experimental results were found. Molecular docking is important to design new molecules with neuroprotective potential. In addition, this powerful tool improves the methodology for finding new molecules. The compound ranking predicts better anti-aggregation properties when the Aβ monomer is used as receptor. Curcumin derivatives showed anti-aggregation and anti-inflammatory properties which suggest that improving the anti-aggregation activity will also improve its anti-inflammatory activity. These molecular modeling studies provide important insights to understanding which chemical interactions are critical to improving the biological activity of therapeutic molecules.
Footnotes
ACKNOWLEDGMENTS
J.L-B, and K.S.R. are grateful to Melo Brain Grant, MEF Nutritional Grant for support and INDICASAT Internal Grant. JL-B. and K.S.R. also thankful to the National Science System (SNI) of National Secretariat for Science, Technology, and Innovation of Panama (SENACYT) for support. J.L-B is grateful to IBRO’s LARC for short stay grant in Universidad Nacional de Colombia. J.A-T thanks DIB-UNAL for financial support. A.O. is grateful to International Brain Research (IBRO) and INDICASAT for the funding to participate in the molecular modeling workshops in Panama City.
M.L.H. is supported by grants from the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute of Ageing (NIA) of the National Institutes of Health (NIH) (award numbers R01NS088645, RF1NS112719, R03AG064266), and Houston Methodist Research Institute funds.
This research was funded by: National Secretariat for Science, Technology, and Innovation of Panama (SENACYT) grant number [FID17–002], INDICASAT Internal Grant [JR04-2020], International Brain Research of Latin America Regional Committee Organization (IBRO’s LARC) for short stay grant in Universidad Nacional de Colombia. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.