
Abstract
Keywords
INTRODUCTION
Frontotemporal dementia (FTD), an important cause of younger-onset dementia characterized by behavior changes and/or language difficulties, may be preceded or followed by the muscle weakness, wasting, and spasticity that is typical of amyotrophic lateral sclerosis (ALS) [1]. Both disorders share a common neuropathological and genetic background [2]. Notably, a hexanucleotide repeat expansion in the C9orf72 gene is the most common known genetic cause of both FTD and ALS [3].
The presence of ALS in FTD heralds a poor prognosis, with a median survival of less than 1.5 years [4, 5]. In addition, FTD patients who develop ALS will benefit from early enrolment in specialized multidisciplinary ALS clinics to optimize care [6], highlighting a need to identify these patients at an early stage.
Longitudinal studies on the development of ALS in FTD have been limited. It has been reported that 10-15% of FTD patients have concomitant ALS at referral [7, 8], but the proportion of patients diagnosed with FTD that will develop ALS over time is less clear. Further, little is currently known about potential clinical predictors of developing ALS in patients presenting with FTD. One study has suggested that psychosis is more common in FTD patients who will develop ALS [9].
This study was therefore designed with two aims: to establish the risk for ALS in a large cohort of longitudinally followed patients with FTD (n = 152), and to determine the clinical variables associated with progression from FTD to FTD-ALS.
MATERIALS AND METHODS
Participants
The study cohort consisted of consecutive patients with FTD referred to the Frontotemporal Dementia Clinical Research Group (FRONTIER) clinic at Neuroscience Research Australia, Sydney, between 2007 and 2016. FTD patients who had concomitant ALS at their first visit were excluded. All included patients were followed prospectively for at least one year.
All participants underwent a comprehensive assessment at each time point, which included a detailed neurological and cognitive examination using a standardized form, neuropsychological evaluation, and structural brain imaging [10]. The assessment procedure incorporates a physical history and clinical neuromuscular examination for bulbar or limb features of ALS. A corroborating and behavioral history was obtained from a close relative or carer of each patient. The family history was reviewed and was considered positive when the patient had at least one first-degree relative with FTD, early onset dementia or ALS (modified Goldman-score of ≤ 3) [11, 12]. The time of onset of cognitive or motor symptoms was collected, mostly from the informant. The diagnosis of probable behavioral variant FTD (bvFTD), progressive non-fluent aphasia (PNFA), or semantic dementia (SD) was made by consensus among a multidisciplinary team according to current clinical diagnostic criteria [13, 14]. Patients with logopenic progressive aphasia were excluded since they almost invariably manifest Alzheimer’s disease pathology [10]. A proportion of patients presented with a mixed picture, that is to say, of prominent behavioral changes meeting the criteria for bvFTD [14] combined with semantic impairment (bvFTD+SD, 14%), or combined with agrammatism or motor speech apraxia, characteristic of PNFA (bvFTD+PNFA, 8%). The Progressive Aphasia Language Scale (PALS) as described before was used to assess language and speech and a score of 2 was considered abnormal [10, 15]. Concomitant ALS was diagnosed when patients also met criteria for probable or definite ALS according to the revised El-Escorial criteria [16]. The diagnosis of ALS was supported by EMG studies in most patients. A neuropathological confirmation of the diagnosis was obtained in 22 patients.
Genomic DNA was obtained for the majority of patients (96%) and was screened for the C9orf72 (NG_031977) hexanucleotide repeat expansion by repeat-primed polymerase chain reaction as described before using genomic DNA [17]. Repeat expansions of more than 30 repeats were considered pathogenic.
Global cognitive function was measured using the Addenbrooke’s Cognitive Examination–III (ACE-III) (range 0–100; scores >88 indicate normal function) [18]. Dementia severity was measured using the Clinical Dementia Rating (CDR) scale [19]. Neuropsychological testing included the span task (forward and backward) of the Wechsler Memory Scale–Third Edition, copy and recall component of the Rey-Osterrieth Complex Figure Test, the FAS Verbal Fluency test, the Sydney Language Battery and the Facial Emotion Selection Task[20–24].
Standard protocol approvals, registrations, and patient consents
Ethical approval was obtained from the ethics committees of the South Eastern Sydney Local Health District and the University of New South Wales. Participants or their responsible surrogates provided written informed consent in accordance with the Declaration of Helsinki.
Statistical analysis
Results of Kolmogorov-Smirnoff tests determined whether variables were normally distributed. We compared parametric variables across groups via independent Student’s t-tests, nonparametric data were analyzed using Mann-Whitney tests and Fisher’s exact tests compared categorical data.
RESULTS
Out of 218 patients with a diagnosis of FTD, 152 were included in the present study. Of those that failed inclusion criteria, 22 (10.1% of total cohort) already had signs of ALS at their first clinical evaluation and 44 had insufficient follow-up data. The mean follow-up was 28.6 months, ranging from 12.0 to 82.8 months. Excluding the patients who developed ALS, the mean length of symptoms at time of last clinical evaluation was 75.6 months, ranging from 21 to 185 months. Retrospective analysis of the 22 patients who had dual FTD-ALS diagnosis at presentation, indicated that the delay between onset of FTD and ALS symptoms usually had been short (mean 22.2 months). The majority of these patients (95.5%) had developed FTD-ALS within 4.5 years from onset of FTD. One had progressed to FTD-ALS after a slowly progressive course with behavioral changes spanning 10 years. Demographic and baseline clinical data of the study cohort are represented in Table 1.
Baseline characteristics of the FTD cohort
mGS, modified Goldman-score; ACE-III, Addenbrooke’s Cognitive Examination third version; CDR, clinical dementia rating scale.
Overall, 8 FTD patients (5.2%) developed ALS. In each patient, symptoms of ALS emerged within one year from the patients’ initial assessment. The mean delay between the onset of FTD symptoms and ALS was 34.5 months (range 19–54 months). We calculated incidence of ALS in the FTD population according to the length of FTD symptoms (Table 2). The incidence of ALS was 6.7 per 100 patient-years in FTD patients with a disease duration of 1 year. A decline was observed in the incidence of ALS in FTD according to the duration of FTD symptoms, particularly when disease duration exceeded 3 years. All patients with FTD-ALS manifested motor features within 5 years from first symptoms of FTD.
Yearly incidence of ALS in FTD patients according to duration of FTD symptoms
Taking FTD subtypes into consideration, of the 11 patients with a mixed bvFTD+PNFA presentation, 5 (45.5%) developed ALS during follow-up (Fig. 1). The risk of developing ALS when presenting with bvFTD+PNFA was markedly higher than with any other FTD phenotype (p < 0.0001, odds ratio 38.3, 95% CI: 7.3 to 199.2). In the bvFTD group, two patients (3.0%) and in the PNFA group, one patient (3.3%) progressed to FTD-ALS. None of the patients seen with an SD or bvFTD+SD phenotype (n = 45) developed ALS subsequently. Excluding those with an SD or bvFTD+SD profile, the rate of ALS development in the cohort was 7.5%.
Of the eight FTD-ALS patients, three had bulbar and limb onset, three had bulbar onset and one, a PNFA patient, had limb onset of ALS symptoms.
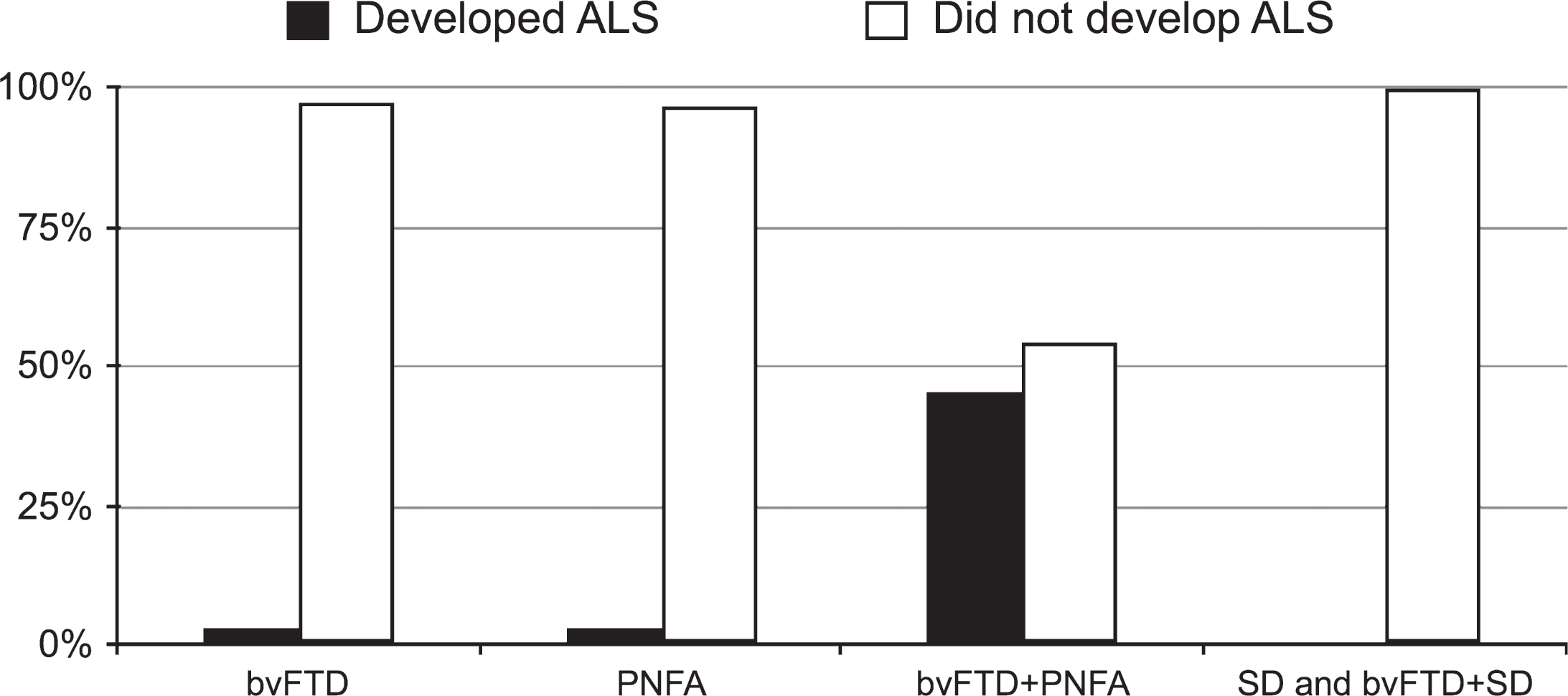
Frequency of development of ALS according to FTD subtypes.
The demographic and clinical features of FTD patients who progressed to FTD-ALS were compared to those without progression to ALS (Table 3). The two groups did not differ with respect to sex distribution and age at onset. Mean survival from disease onset in 6 deceased FTD-ALS patients was significantly shorter than in patients with FTD who did not develop ALS (34.5 versus 101.4 months, p < 0.04). The proportion of patients with a positive family history was similar between groups. FTD patients who developed ALS more often carried the C9orf72 repeat expansion than those who did not (p = 0.02, odds ratio 8.0, 95% CI: 1.7 to 38.6). Two out of 3 FTD patients who carried the C9orf72 repeat expansion and developed ALS had no know family history of a neurodegenerative disorder. Further, the patients who progressed to FTD-ALS exhibited more agrammatism (p = 0.01, odds ratio 7.2, 95% CI: 1.6 to 32.0) and apraxia of speech (p = 0.03, odds ratio 5, 95% CI: 1.1 to 22.0) at baseline assessment. Of note, none of the FTD patients who developed ALS had presented with psychotic symptoms. The neuropsychological data showed that FTD patients who developed ALS performed significantly worse on the single word repetition task of the SYDBAT language battery (p = 0.006) and the FAS verbal fluency task (p = 0.02), indicating a more pronounced language deficit. The FTD patients who developed ALS also performed worse on a facial emotion selection task (p = 0.04). In contrast, the two groups did not differ on measures of general cognition (ACE-III), attention (Digit Span), visuospatial ability (Rey Complex Figure copy), visual memory (Ray Complex Figure recall), naming or comprehension (naming or comprehension subtests of the SYDBAT).
Clinical characteristics of FTD patients who developed ALS compared with those that did not develop ALS
ACE-III, Addenbrooke’s Cognitive Examination third version; RCF, Rey Complex Figure; SYDBAT, Sydney Language Battery
When the clinical characteristics of five bvFTD+PNFA patients who developed ALS were compared to six bvFTD+PNFA patients who did not, no significant differences were found (Supplementary Table 1). The mean length of FTD symptoms in the bvFTD+PNFA without ALS group was 60.8 months (range 36–104 months). Two patients in this group (with shortest clinical follow-up) have come to autopsy. Both had FTLD-TDP type of pathology without evidence of ALS.
DISCUSSION
Overall, eight (5%) FTD patients developed ALS on follow-up. The longitudinal data indicated that the risk to develop ALS declined with the length of FTD symptoms. The incidence of ALS was 6.7 per 100 patient-years in FTD patients with a disease duration of one year. Lower rates were found thereafter, and after a disease duration of 5 years, no FTD patient developed ALS. This is consistent with prior descriptions which indicated that the delay between the onset of FTD and ALS is usually short [5, 26]. One previous study that included 43 FTD-ALS patients with FTD onset before ALS, reported a median delay of 19 months and that over 90% of patients had developed the motor symptoms by an FTD disease duration of more than 4 years [5]. Nonetheless, FTD patients with an onset of ALS 10 years after the first manifestation of behavioral changes have been reported [27]. Such a late onset of ALS symptoms in an FTD patient, however, appears to be a very rare event.
The frequency of ALS in FTD has not been fully characterized. Two cross-sectional studies have indicated that about 10–15% of FTD patients who are referred to a cognitive disorders clinic also have concomitant ALS [7, 8]. Another study found that no FTD patients progressed to FTD-ALS during a follow-up period of about 2.5 years, but this was based on a modest number of 38 patients [28]. In our own experience of 215 patients with an FTD diagnosis (excluding logopenic progressive aphasia), 22 had concomitant FTD-ALS at presentation while a further 8 developed ALS at follow-up, giving an overall prevalence of 14.0%. This indicates that a presentation with features of both disorders is a common occurrence.
A key finding of this study is that the bvFTD+PNFA phenotype is strongly associated with increased risk for progression to FTD-ALS. A substantial proportion of bvFTD patients develop a degree of semantic impairment or show a reduction of in spontaneous speech (adynamic speech) [29, 30], but frank motor speech disturbance and/or agrammatism sufficient to qualify a dual diagnosis is rare and was found in only 7.2% (11 of 152) patients. When followed longitudinally, almost half of those bvFTD+PNFA patients progressed to develop ALS, making this a high-risk state for the development of ALS (38-fold increased risk compared to other FTD phenotypes). The constellation of behavioral abnormalities, non-fluent aphasia, and ALS has been described before [31–33]. For instance, Bak et al. (2001) reported six patients with ALS and early progressive non-fluent aphasia, of which four also showed a marked change in personality [33]. Recent work using the Test for the Reception of Grammar (TROG) has also found that impaired comprehension of syntactically complex sentences, which is typically present in PNFA, is also prevalent in patients with FTD-ALS [34].The association between the combination of bvFTD and PNFA and the subsequent development of ALS, however, has not been documented previously.
Patients classified as PNFA had motor speech apraxia and/or agrammatism. All underwent a thorough neurological examination and did not show clinical evidence for bulbar palsy at baseline. Yet, the majority of FTD patients who progressed to develop ALS later manifested bulbar symptoms of ALS. Previous studies have also reported a higher prevalence of bulbar onset in patients with FTD-ALS compared to ALS without dementia [26, 35]. The explanation for this phenomenon remains unclear. It has been suggested that regional spread of pathology might be relevant because the peri-insular region, which is characteristically affected in PNFA, is located close to bulbar motor cortical regions [36].
It should also be noted that despite shared TDP-43 pathology no patients with SD developed ALS, in keeping with prior studies [37]. The TDP-43 pathology in SD has a distinct pattern, designated FTLD-TDP type C, which differs from that found in ALS and patients with bvFTD or PNFA who show TDP-43 pathology [38], suggesting differences in the underlying disease mechanisms.
Besides the identified risk phenotype, FTD patients who carried the C9orf72 repeat expansion were also more likely to develop ALS (8-fold increased risk). This is in keeping with reports of a higher C9orf72 repeat expansion frequency in FTD-ALS patients compared to FTD patients without ALS [3]. Of note is that, similar to as previously reported, the majority of FTD patients who developed ALS did not carry the C9orf72 repeat expansion [3, 26]. Therefore, the presence of a C9orf72 repeat expansion to predict progression to FTD-ALS has a low sensitivity.
We could not confirm that psychosis is a reliable predictor of FTD-ALS in a patient presenting with FTD. The overall frequency of psychosis in FTD-ALS patients as reported in the literature is relatively low (10–15%) [25, 26]. A recent study that compared the clinical phenotype of FTD-ALS patients with and without C9orf72 repeat expansions found that psychotic symptoms commonly occurred in those with the expansion (65%) (n = 11) but were rare (5%) in 49 FTD-ALS patients without the expansion [26]. Similarly, several studies have suggested that psychosis is a marker for C9orf72 repeat expansions in FTD patients without ALS [39, 40]. Thus, the association between FTD-ALS and psychosis as described before [9], is probably explained by differences in C9orf72 repeat expansion frequency between FTD and FTD-ALS groups, and not by ALS itself.
Some caveats should be kept in mind when interpreting our results. Our findings are derived from a clinic-based cohort, which might not reflect the general FTD population. To further delineate the prevalence and incidence of ALS in FTD patients, prospective population-based studies will be needed. Although our patient groups were well characterized, pathological confirmation of the diagnosis was not available for the majority of patients. In addition, it is possible that some FTD patients might develop ALS when followed over a longer time period. We expect, however, that this is a rare event, given that 81% have been followed for longer than 4 years from FTD onset.
In summary, we identified two risk factors that are associated with development of ALS in FTD patients, a mixed bvFTD+PNFA phenotype and the presence of a C9orf72 repeat expansion. This observation has important implications for clinical practice. Close monitoring of such patients offers the opportunity to identify ALS at an early stage. If ALS emerges, the patient could benefit from early administration of disease-modifying drugs and referral to a multidisciplinary ALS clinic, which has been shown to improve outcomes in the management of ALS [6]. In addition, our results may allow improved prediction of risk to develop ALS in a patient who is diagnosed with FTD, which appears to decline with length of FTD symptoms.
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge Barbara Toson for her comments on statistics methods conducted in this report. This work was supported by funding to Forefront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neuron disease, from the National Health and Medical Research Council of Australia program grant (#037746) and the Australian Research Council Centre of Excellence in Cognition and its Disorders Memory Program (#110001021). In addition, OP is supported by an NHMRC Senior Research Fellowship (#1103258), JRB by an NHMRC Early Career Fellowship (#1072451), CL by an NHMRC-ARC Dementia Research Development Fellowship (#1102969), JBK by an NHMRC Dementia Research Team Grant (#1095127) and GH by an NHMRC Senior Research Fellowship (#1079679).