
Abstract
Background:
Abnormally hyperphosphorylated tau is the major protein of neurofibrillary tangles in Alzheimer’s disease. Insulin activates PI3K-AKT signaling and regulates tau phosphorylation. Impaired brain insulin signaling is involved in Alzheimer’s disease pathogenesis. However, the effect of peripheral insulin on tau phosphorylation is controversial.
Objective:
In the present study, we determined the effect of peripheral insulin administration on tau phosphorylation in brain.
Methods:
We intraperitoneally injected a super physiological dose of insulin to mice and analyzed PI3K-AKT signaling and tau phosphorylation in brains by western blots.
Results:
We found that peripherally administered insulin activated the PI3K-AKT signaling pathway immediately in the liver, but not in the brain. Tau phosphorylation in the mouse brain was found to be first decreased (15 min) and then increased (30 min and 60 min) after peripheral insulin administration and these changes correlated inversely with body temperature and the level of brain protein O-GlcNAcylation. Maintaining body temperature of mice post peripheral insulin administration prevented the insulin/hypoglycemia-induced tau hyperphosphorylation after peripheral insulin administration.
Conclusion:
These findings suggest that peripheral insulin can induce tau hyperphosphorylation through both hypothermia and downregulation of brain protein O-GlcNAcylation during hypoglycemia.
INTRODUCTION
Insulin is the main peptide hormone secreted by β cells of the pancreas islets. It promotes the uptake of glucose by liver, fat, and skeletal muscle cells from the blood and regulates the metabolism of carbohydrates, fats, and proteins [1, 2]. Insulin’s functions are mainly mediated through the phosphoinositide-3 kinase (PI3K)–v-AKT (murine thymoma viral oncogene homolog) and the mitogen activated kinase (MAPK) pathways [2]. The brain has long been considered as an insulin-insensitive because brain glucose uptake is not affected by insulin [3, 4]. Over the last decades, accumulating evidence suggests that the brain is an important target for insulin action [5, 6]. Insulin in the brain regulates feeding behavior and body energy stores, the metabolism of glucose and fats in the liver and adipose, and various aspects of memory and cognition [6]. Disturbances of cerebral insulin signaling have been implicated in the pathophysiology of brain ageing and dementia [7, 8].
Alzheimer’s disease (AD) is the major form of dementia in the aged population. Neurofibrillary tangles, aggregates of abnormally hyperphosphorylated tau, are a hallmark of this disease. Tau is a major neuronal microtubule binding protein. The major function of tau is to stimulate microtubule assembly and to stabilize microtubule network. In AD brain, tau is abnormally hyperphosphorylated [9, 10]. Hyperphosphorylation of tau is believed to be responsible for the loss of its biological function and the gain of toxicity and aggregation [11, 12]. Glycogen synthase kinase-3β (GSK-3β), a key downstream kinase of AKT, is a major tau kinase and it phosphorylates tau at multiple sites [13 –16]. Thus, insulin signaling modulates phosphorylation of tau protein. Impairment of the PI3K-AKT pathway in AD brain may be involved in the development of this disease [17 –19].
Although the presence of insulin in the cerebrospinal fluid (CSF) is well documented, the origin of brain insulin is controversial [20]. Studies using exogenous or radioactively labeled insulin indicated that insulin crosses the brain-blood barrier (BBB) by a saturable mechanism, but transport of insulin to the brain via CSF is slow [21, 22]. It was reported that peripheral administration of insulin transiently increases the phosphorylation of AKT and GSK-3β in mouse brain [23]. However, peripheral hyperinsulinemia was reported to have no substantial influence on the phosphorylation of AKT, GSK-3β or tau in the mouse brain [24]. Thus, functions of peripheral insulin in central nervous system remain elusive.
To determine the effect of peripheral insulin on the PI3K-AKT pathway and tau phosphorylation in the brain, we peripherally administrated a super physiological dose of insulin (5 I.U./kg) to mice and analyzed PI3K-AKT signaling and tau phosphorylation in mouse brains. We found immediate activation of the PI3K-AKT signaling pathway in the liver but not in the brain. Tau phosphorylation was found to be first decreased (15 min) and then increased (30 and 60 min) after peripheral insulin administration, and was correlated to hypothermia and decreased O-GlcNAcylation of global proteins.
MATERIALS AND METHODS
Animals
Male C57BL/6J mice (5–6 months of age) obtained from the Experimental Animal Center of Nantong University (Nantong, Jiangsu, China) were employed for this study. The mice were housed with a 12 h/12 h light/dark cycle and with ad libitum access to food and water. All animal experiments were approved by Institutional Animal Care and Use Committee of Nantong University, according to the United States PHS Policy on Human Care and Use of Laboratory animals.
Antibodies and reagents
Primary antibodies used in this study are listed in Table 1. Peroxidase-conjugated anti-mouse and anti-rabbit IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The enhanced chemiluminescence (ECL) kit was from Pierce (Rockford, IL, USA). Humulin R (U-100) was from Eli Lilly Company (Indianapolis, IN, USA). Other chemicals were from Sigma (St. Louis, MO, USA).
Antibodies used in the present study
Mono-, monoclonal; p-, phosphorylated; up-, unphosphorylated; Poly-, polyclonal; M, Mouse; R, Rabbit.
Intraperitoneal injection of insulin
Mice were first habituated to handling for 7 consecutive days before they were used for the study. After 2-h fasting, the body weight, body temperature and blood glucose level (as 0 time point) were measured and then insulin (5 I.U./kg) was administrated by intraperitoneal injection. The body temperature and blood glucose level were measured at 5, 15, 30, and 60 min after insulin administration, followed by immediate sacrifice of mice by cervical dislocation. The cerebral cortex, hippocampus, and liver were immediately dissected and stored at –80°C until used. Control mice (or 0 min) were sacrificed after measuring body temperature and blood glucose without insulin intraperitoneal injection (control group, n = 5; 5 min, n = 6; 15 min, n = 7; 30 min, n = 6; 60 min, n = 6). Saline injection was used for control.
Measurement of body temperature
During the temperature acquisition, the base of the tail was fixed with two fingers and then gently lifted while the animal gripped a metal rod on the cage lid with its front paws, thus allowing for exposure of the ano-genital area. Non-contact infrared thermometers (Non-Contact Thermometer, Walgreens) were used to measure the perianal regions temperature.
Measurement of blood glucose
Blood samples (∼3μl) were collected from tail tip at 0, 5, 15, 30, and 60 min after insulin administration by a skilled technician. The blood glucose levels were measured using a Glucometer (Bayer Contour, Tarrytown, NY, USA). Mice were kept free during tail-tip amputation other than fixed by a retainer or held by hand.
Western blot analysis
Mouse left cortex and liver were homogenized in pre-cooled buffer containing 50 mM Tris-HCl (pH7.4), 2 mM EDTA, 2 mM EGTA, 150 mM NaCl, 50 mM NaF, 1.0 mM Na3VO4, 10 ug/ml aprotinin, 10μg/ml leupeptin, 10μg/ml pepstatin, and 1.0 mM 4-(2-aminsoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF). The homogenate was immediately mixed with the same volume of 2 x Lamaeli buffer (125 mM Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 2% 2-mercaptoethanol, and 40% bromophenal blue), followed by heating in boiling water for 5 min. The protein concentration was determined by using the A660 kit (Pierce Rockford, IL) according to the manufacturer’s instructions. The samples were resolved in 10% SDS-PAGE and electro-transferred onto Immobilon-P membrane (Millipore, Bedford, MA, USA). The blots were then probed with primary antibody and developed with the corresponding horseradish peroxidase-conjugated secondary antibody and enhanced chemiluminescence kit (ECL, Pierce Biotechnology, Rockford, IL). Densitometric quantification of protein bands in western blots were analyzed by using the Multi Gauge V3.0 software (Fuji Photo Film Co., Ltd).
Statistical analysis
GraphPad Prism 6 was used for statistical analysis. Results were analyzed by one-way ANOVA for multiple-group analysis followed with Tukey’s multiple comparisons test. To study the association, Pearson correlation analysis was performed. p < 0.05 was considered statistically significant.
RESULTS
Peripherally administrated insulin activates PI3K-AKT signaling in the liver, but not in the brain in mice
The PI3K-AKT signaling pathway is the major respondent of insulin stimulation. Activation of insulin receptor upon binding to insulin activates the PI3K-AKT signaling pathway through tyrosine phosphorylation of the p85 regulatory subunit of PI3K, serine phosphorylation of 3-phosphoinositide-dependent protein kinase-1 (PDK1), serine/threonine phosphorylation of AKT, and serine phosphorylation of GSK-3β in tandem. To assess the activation, we measured the phosphorylation of these components of the PI3K signaling pathway in the mouse tissue by using quantitative western blots. We found that levels of phosphorylated PI3K at Tyr458, AKT at Ser473, and GSK-3β at Ser9 were increased in the liver in a time-dependent manner (Fig. 1), suggesting a rapid activation of the PI3K-AKT pathway in liver after peripheral insulin administration.

Activation of PI3K-AKT signaling in the liver after intraperitoneal injection of insulin in mice. C57BL/6 mice were injected intraperitoneally with insulin and scarified at 5, 15, 30, and 60 min after injection. The phosphorylation levels of proteins in the PI3K-AKT pathway in right hepatic lobule were analyzed by western blots developed with indicated antibodies (A) and normalized with the level of corresponding protein after density quantification and are shown as mean±SEM (B). *p < 0.05, ***p < 0.001, ****p < 0.0001.
Whether peripheral insulin crosses blood-brain-barrier and works in the central nervous system is controversial. To identify the role of peripheral insulin in the brain, we analyzed the phosphorylated proteins involved in PI3K-AKT signaling pathway in the brains of mice after peripheral administration of insulin. We did not find similar time-dependent phosphorylation of PI3K, PDK1, AKT, or GSK-3β, the major components of the PI3K-AKT signaling pathway in the brain as we found in the liver upon insulin administration (Fig. 2). Instead, we found a moderate reduction of phosphorylation of three out of the four kinases studied during the first 15 min, followed by elevation of the phosphorylation at longer time points in the brain after peripheral insulin administration. These results suggest that peripherally administrated insulin fails to act in the brain directly as it did in the liver. To verify this phenomenon, we determined the phosphorylation of other downstream components of insulin signaling, including tuberous sclerosis complex 2 (TSC2) at Thr1462, mammalian target of rapamycin (mTOR) at Ser2448, 5′ AMP-activated protein kinase (AMPK) at Thr172, and extracellular signal–regulated kinase (ERK) at Thr202/Tyr204. We did not find immediate phosphorylation of any of these kinases in the brain upon peripheral insulin administration (Fig. 2). Taken together, these results indicate that peripherally administrated insulin does not act in the brain.
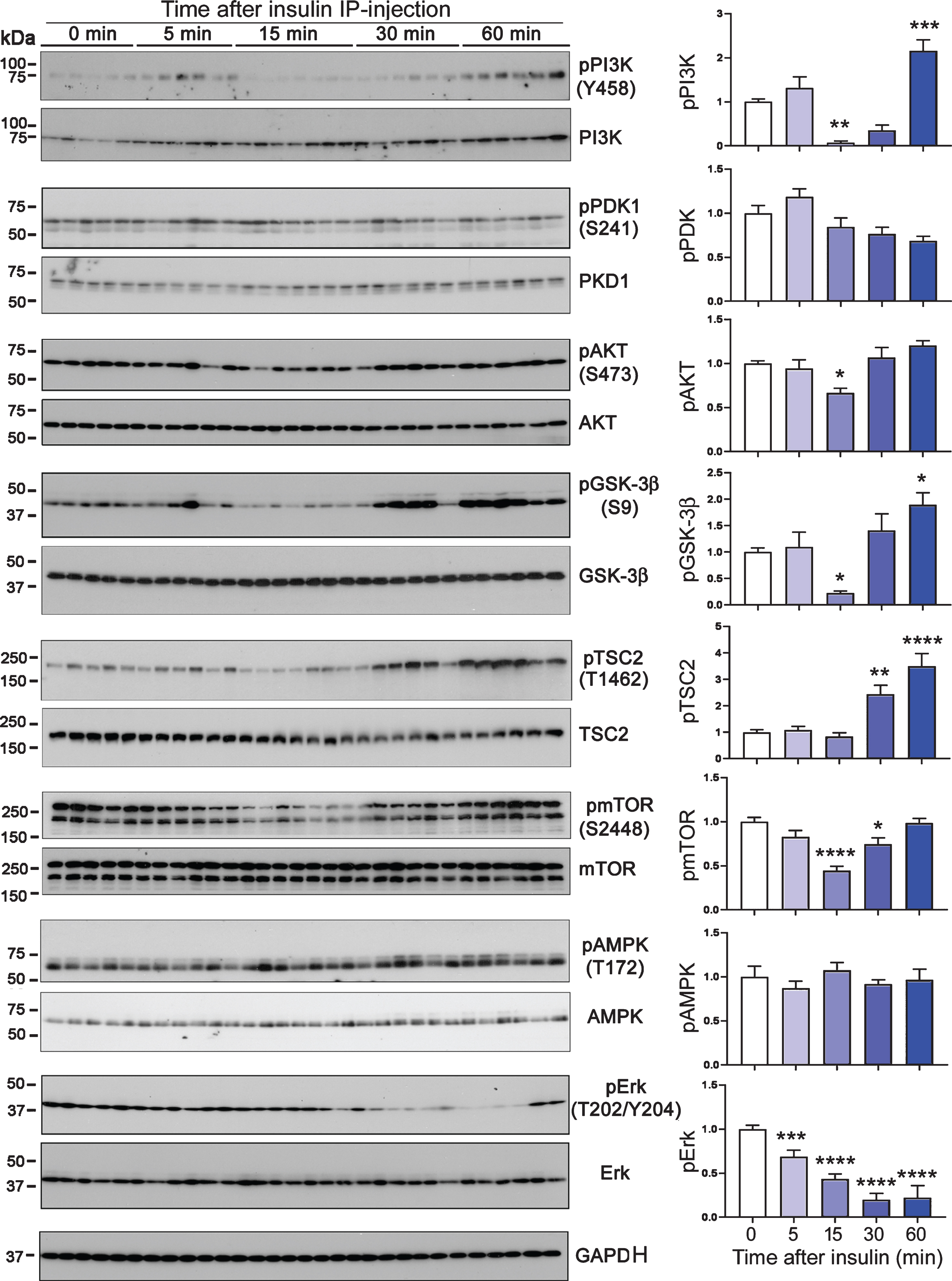
Phosphorylation of common insulin respondent proteins in the mouse brains after peripheral insulin administration. Phosphorylation levels of the indicated proteins were analyzed by western blots. The graphs show the quantifications after normalization with the corresponding kinase and are presented as mean±SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Peripheral insulin administration correlates with tau phosphorylation site-specifically in the brain
To study if peripherally administrated insulin has any impact on tau phosphorylation in the brain, we analyzed phosphorylation of tau by western blots developed with site-specific and phosphorylation-dependent tau antibodies. We found that the phosphorylation levels of tau at Ser199, Thr205, Thr212, Ser214, Thr217, Ser262, Ser396, and Ser404 were slightly reduced or showed a trend to reduce 15 min after insulin administration (Fig. 3). Following the slight reduction, tau phosphorylation levels were clearly increased or showed trend to increase 30 min and 60 min after peripheral insulin administration (Fig. 3). It is interesting to note that these dynamic changes of tau phosphorylation corresponded to changes of most of brain kinases studied above (see Fig. 2), but not to those of liver kinases (see Fig. 1).
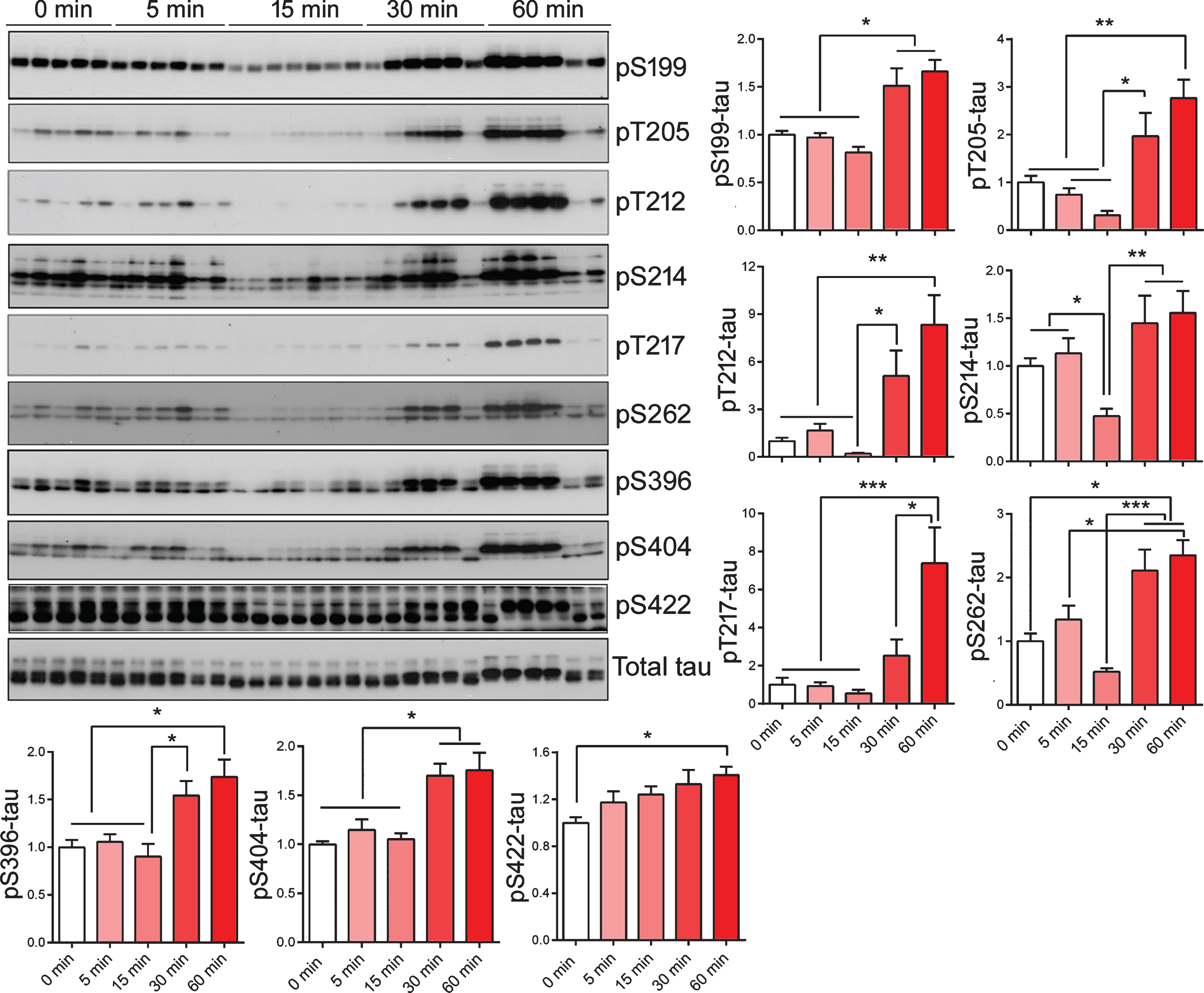
Alterations of tau phosphorylation in brains of mice after peripheral insulin administration. The phosphorylation of tau in the mouse brains after intraperitoneal injection of insulin was analyzed by western blots developed with the indicated site-specific tau antibodies. The level of phosphorylated tau at individual sites were normalized to total tau (R134d) and are shown as mean±SEM in graphs. *p < 0.05, **p < 0.01, ***p < 0.001.
Peripheral insulin administration may promote tau phosphorylation in the mouse brain through hypothermia associated with insulin-induced hypoglycemia
Correspondence between changes in phosphorylation/activation of some of the kinases shown in Fig. 2 and tau phosphorylation levels shown in Fig. 3 suggest that these kinases could contribute to the dynamic alterations of tau phosphorylation after peripheral insulin administration because these kinases could phosphorylate tau at multiple sites [25 –27]. However, the phosphorylation of GSK-3β at Ser9 inhibits rather than activates its kinase activity [28], which eliminates the possibility of the involvement of GSK-3β to tau phosphorylation under this condition. Otherwise, tau phosphorylation would have increased at 5 min and decreased at 15–30 min post peripheral insulin administration. To investigate the possible mechanism that leads to the dynamic changes of tau phosphorylation in the brain following peripheral insulin administration, we determined the body temperature of the mice post peripheral insulin administration because insulin can induce hypoglycemia and in turn hypothermia [29] that has been shown to induce tau phosphorylation [30 –32]. We found the time-dependent reduction of body temperature as well as the blood glucose level in mice post peripheral insulin administration (Fig. 4A–D). Correlation analyses between the body temperature and tau phosphorylation at the specific phosphorylation sites indicated inverse correlation at all the phosphorylation sites studied (Fig. 4E). These results suggest that tau hyperphosphorylation found 30–60 min post peripheral insulin administration could be caused by marked hypoglycemia-induced hypothermia.

Inverse correlation between hyperphosphorylation of tau and hypothermia in the brains of mice with peripheral insulin administration. A-D) Blood glucose (A) and body temperature (B) were measured at various time point after insulin IP-injection and are presented as mean±SEM. Correlation between hypothermia and blood glucose level in mice administrated with insulin up to 60 min (C) or 30 and 60 min (D) were analyzed by non-linear regression. E) Correlation of body temperature and phosphorylation of tau at individual sites in mice with IP-injection of insulin for 30 and 60 min was analyzed by Pearson correlation analysis.
It was previously reported that hypothermia can modulate protein phosphatase 2A (PP2A) through methylation of its catalytic subunit [30, 32], which is a major brain phosphatase regulating tau phosphorylation [33]. We therefore determined PP2A catalytic subunit (PP2Ac) demethylation at leucine 309, which inhibits PP2A activity by affecting its association with regulatory B subunits [34 –36]. We found that the level of demethylated PP2Ac was increased in mouse brains at 5 and 15 min after insulin administration and returned to basal level 30 and 60 min after insulin injection (Fig. 5A,B). These results suggest a transient inhibition of PP2A activity during 5–15 min post peripheral insulin administration, which cannot explain the increased tau phosphorylation occurred 30–60 min post insulin administration.

Alteration of PP2Ac demethylation in the brain in insulin IP-injected mice. Demethylation of PP2A catalytic subunit at Leu309 (DM-PP2Ac) was analyzed by western blots (A) in brains of mice administrated peripherally with insulin and normalized with PP2Ac and are presented as mean±SEM (B).
Maintaining body temperature prevents hyperphosphorylation of tau induced by peripheral insulin administration
To verify the role of hypothermia in the increased level of tau hyperphosphorylation post peripheral insulin administration, we added a control group for which the body temperature of the mice was maintained at 37°C by putting the animals on a warm pad post insulin administration. As above, we found a moderate decrease at 15 min, followed by elevation of tau hyperphosphorylation at 30 and 60 min post intraperitoneal insulin administration in mice (Fig. 6A). When the body temperature was maintained post insulin administration, the insulin-induced increase in tau hyperphosphorylation was prevented completely (Fig. 6A,D). These results indicate that tau hyperphosphorylation found 30 min post peripheral insulin administration was caused by insulin-induced hypothermia. In addition to tau hyperphosphorylation, the phosphorylation of AKT at Ser473 and GSK-3β at Ser9 was also increased at 30–60 min post peripheral insulin administration (see Fig. 2). We, thus studied whether the increased phosphorylation of these two kinases was also caused by hypothermia. We found that, as for AKT and GSK-3β phosphorylation, the phosphorylation of these two kinases was prevented completely if the body temperature of the mice was maintained post peripheral insulin administration (Fig. 6B,C). Taken together, these results suggest that hypothermia underlies hyperphosphorylation of tau and some protein kinases like AKT and GSK-3β in the brain after peripheral administration of insulin.

Reversal of insulin-induced hyperphosphorylation of AKT, GSK-3β and tau by heat preservation. C57BL/6 mice were injected intraperitoneally with insulin, heat preserved at 37°C or room temperature and scarified at 15, 30, and 60 min after injection. The phosphorylation levels of AKT, GSK-3β and tau were analyzed by western blots developed with indicated antibodies (A), normalized with the corresponding protein and are presented as mean±SEM (B–D). *p < 0.05, **p < 0.01, ***p < 0.001.
O-GlcNAcylation correlates negatively with phosphorylation of tau in brains of mice after peripheral insulin administration
Tau protein is also modified by O-GlcNAcylation, a post-translational modification in which the monosaccharide-N-acetyl-glucosamine (GlcNAc) attaches to serine/threonine residues of a protein [37, 38]. O-GlcNAcylation negatively regulates tau phosphorylation [9, 34]. We previously found that fasting-induced hypoglycemia can lead to decreased O-GlcNAcylation and increased phosphorylation of tau in brain [38, 39]. We, thus studied the effect of peripheral insulin administration on protein O-GlcNAcylation in mouse brain by using western blots developed with RL2, an antibody that binds only to O-GlcNAc-modified proteins. We found decreased protein O-GlcNAcylation in mouse brains 30–60 min post peripheral insulin administration (Fig. 7A, B), which coincided with increased tau phosphorylation during the same time periods (see Fig. 3). Correlation analyses indicated positive relationship between blood glucose levels and global protein O-GlcNAcylation (Fig. 7C) and inverse relationship between global protein O-GlcNAcylation and tau phosphorylation at most of the sites studied (Fig. 7D). These results suggest that peripheral insulin may also induce tau hyperphosphorylation in the brain through down-regulation of brain protein O-GlcNAcylation as a result of hypoglycemia. This confirmed our previous findings of the reciprocal regulation between O-GlcNAcylation and phosphorylation of tau in the brain.

Association of reduced O-GlcNAcylation with phosphorylation of tau in brains of mice with peripheral insulin administration. A) Protein O-GlcNAcylation in brains of insulin-injected mice was analyzed by western blots developed with RL2, normalized by GAPDH and are presented as mean±SEM (B). *p < 0.05. C) Correlation between blood glucose and O-GlcNAcylation in brain of insulin injected mice was analyzed by Pearson linear regression. D) Correlation between O-GlcNAc and phosphorylation of tau in brains of mice was analyzed by Pearson correlation analysis.
DISCUSSION
Abnormal hyperphosphorylation of tau is critical in AD pathogenesis. Insulin signal regulates tau phosphorylation [19, 40], but the role of peripheral insulin in the central nervous system is controversial. In the present study, we found that peripherally administrated insulin activated the insulin signaling, as determined by activation of the PI3K-AKT signaling pathway, in the liver immediately, but did not affect insulin signaling in the brain in mice directly. Delayed increase in tau phosphorylation at multiple phosphorylation sites was observed in the brain after peripheral insulin administration, and it was correlated to the insulin/hypoglycemia-induced hypothermia and brain protein O-GlcNAcylation. Our findings suggest that probably intraperitoneally administered insulin does not activate PI3K signaling in the brain and that tau hyperphosphorylation can be induced through both hypothermia and downregulation of brain protein O-GlcNAcylation during hypoglycemia.
Insulin has emerged as a major regulator within the central nervous system. Whether insulin crosses BBB was debated for decades. As early as 1954, two studies examined the ability of peripherally injected radio-labeled insulin to enter the brain and concluded that insulin did not cross the BBB [41, 42], which was confirmed later by several other studies in mid 1980 s [5, 43]. However, several groups found a correlation of the level of insulin between the cerebrospinal fluid (CSF) and serum and that this correlation flattened as insulin levels increased out of the physiologic range, raising the possibility that insulin could cross the BBB by a saturable mechanism [44 –46]. However, this belief was counterargued by that CSF insulin was derived from CNS sources and not from the blood [5 , 47]. Later studies using exogenous or radioactively labeled insulin confirmed that insulin crossed the BBB by a saturable mechanism [21 , 48–50]. The concentration of insulin in plasma is 10- to 20-fold higher in healthy individuals and transport of insulin to the brain via the CSF circulation is slow [51, 52]. Chronic peripheral hyperinsulinemia has no substantial influence on phosphorylation of AKT and GSK-3β [24]. It was reported that administration of 5 I.U./kg insulin caused transient increase in phosphorylation of AKT and GSK-3β 5 min after insulin administration in cerebral cortex and hippocampus, which rapidly reverted to basal levels, although the insulin concentration remained elevated [23]. However, in the present study by IP-injection of 5 I.U./kg insulin, instead of quick activation we found downregulation of PI3K-AKT pathway such as decreased phosphorylation of GSK-3β and other proteins of PI3K pathway in mouse brains 5 min and 15 min after the injection. However, the phosphorylation of these proteins of PI3K pathway was increased 30 min later after insulin administration, which was associated with hypothermia. Thus, brain PI3K signaling was not activated by peripheral administration of super physiological dose of insulin, indicating that no effective level of insulin crosses BBB and thus, peripheral insulin is unlikely to function to activate insulin pathways in the brain.
Reduction of blood glucose is associated with the hypothermia, which causes tau hyperphosphorylation. Tau is hyperphosphorylated in the brain of hibernating animals [53 –55]. In the hibernating animal brain, GSK-3β phosphorylation at Ser9 is increased [56]. Anesthesia causes hypothermia, resulting in tau hyperphosphorylation [31, 32]. In the present study we found that peripheral insulin administration induced hypothermia and increased the phosphorylation of GSK-3β and tau. We speculate that hypothermia is involved in peripheral insulin induced tau hyperphosphorylation. Interestingly, peripheral insulin administration did not induce Ser422 hyperphosphorylation. Ser422 is phosphorylated by Erk1/2 and SAPK and dephosphorylated by PP2A and PP2B dephosphorylate [57]. Similarly, Ser422 is not hyperphosphorylated in anesthetic mouse brain [31], but it is abnormally hyperphosphorylated in AD brain [39], not rapidly dephosphorylated as tau at other phospho-sites during postmortem period [58]. Moreover, we recently found that Ser422 is abnormally hyperphosphorylated in both ipsilateral and contralateral hippocampi of Tg/hTau mice injected with pathological tau from AD brain [59]. Thus, Ser422 hyperphosphorylation may be critical in tau pathogenesis. In addition to phosphorylation, tau is modified by O-GlcNAcylation [37, 38], which negatively regulates tau phosphorylation in site-specific manner [38, 39]. The activity of O-GlcNAc transferase is sensitive to relatively small changes in UDP-GlcNAc availability over a wide range of concentrations [60]. Intracellular glucose level affects O-GlcNAcylation via UDP-GlcNAc generated from glucose. In AD brain, O-GlcNAcylation is reduced and negatively correlated to tau phosphorylation due to impairment of glucose uptake [39]. We here found that O-GlcNAcylation in brain was decreased in time dependent of insulin-injection and coincided with blood glucose decrease. Thus, decreased O-GlcNAcylation might also contribute to tau hyperphosphorylation in the brain of mice 30 min and 60 min after insulin treatment.
Interestingly, total tau level was a bit lower in 15 min at RT and 30 min at 37°C groups compared with corresponding others. Tau in these two groups was less phosphorylated. It was believed that increased phosphorylation of tau might reduce tau degradation [61, 62]. Thus, the slight decrease of tau level might result from increased degradation. In addition, we found that phosphorylation of tau, AKT, GSK-3β, and mTor in mouse brains was decreased at 15 min post insulin administration. At this time point, body temperature and O-GlcNAcylation level were all not significantly altered, but glucose level started to decrease. We previous found that tau, AKT, and GSK-3β were rapidly dephosphorylated after death [58], which may share similar mechanism by reduction or stop metabolism caused by insulin or lower blood glucose, which, however, remains to determined.
PP2A and GSK-3β are the major modulators of tau phosphorylation. Methylation of PP2Ac enhances its activity to dephosphorylate tau [63]. In addition to methylation, PP2A activity is greatly affected by the temperature [30]. PP2A and GSK-3β regulate each other. PP2A dephosphorylates GSK-3β and GSK-3β modulates PP2Ac demethylation by inhibiting the expression of protein phosphatase methylesterase-1 [63, 64]. Since dephosphorylation of GSK-3β increases its activity and demethylation of PP2A suppress inhibits its activity, decreased tau phosphorylation at 15 min may not result from GSK-3β or/and PP2A. However, the molecular mechanism involved in the reduction of Ser9 phosphorylation of GSK-3β and elevation of DM-PP2Ac at 15 min after insulin injection is unknown. In addition to methylation, PP2A activity is highly regulated by temperature [32]. Thus, at 30- and 60-min post-insulin administration, decreased PP2A activity induced by hypothermia probably causes increase in inhibitory phosphorylation of GSK-3β at Ser9. The hyperphosphorylation of tau at these two time points lead us to speculate that compared to the effect of hypothermia and O-GlcNAylation, the impact of the inhibition of GSK-3β in tau phosphorylation might be minimal. As a major tau phosphatase, decrease of PP2A activity caused by hypothermia might be critical in tau hyperphosphorylation induced by peripheral administration of insulin.
In summary, peripheral insulin might not function to activate insulin pathways and to suppress tau hyperphosphorylation in the brain directly, but instead modulate signaling of PI3K-AKT and induce tau hyperphosphorylation through hypothermia and decrease in O-GlcNAcylation. These findings provide an explanation on how peripheral insulin can contribute to tau pathology.
Footnotes
ACKNOWLEDGMENTS
This work was supported in part by Nantong University and New York State Office for People with Developmental Disabilities and by grants from the U.S. Alzheimer’s Association (Grant DSAD-15-363172) and the Neural Regeneration Co-innovation Center of Jiangsu Province.