
Abstract
Background:
Studies have demonstrated that both tau and cardiovascular risk are associated with cognitive decline, but the possible synergistic effects of these pathologic markers remain unclear.
Objective:
To explore the interaction of AD biomarkers with a specific vascular risk marker (pulse pressure) on longitudinal cognition.
Methods:
Participants included 139 older adults from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Biomarkers of tau, amyloid-β (Aβ), and vascular risk (pulse pressure) were assessed. Neuropsychological assessment provided memory, language, and executive function domain composite scores at baseline and 1-year follow-up. Multiple linear regression examined interactive effects of pulse pressure with tau PET independent of Aβ PET and Aβ PET independent of tau PET on baseline and 1-year cognitive outcomes.
Results:
The interaction between pulse pressure and tau PET significantly predicted 1-year memory performance such that the combined effect of high pulse pressure and high tau PET levels was associated with lower memory at follow-up but not at baseline. In contrast, Aβ PET did not significantly interact with pulse pressure to predict baseline or 1-year outcomes in any cognitive domain. Main effects revealed a significant effect of tau PET on memory, and no significant effects of Aβ PET or pulse pressure on any cognitive domain.
Conclusion:
Results indicate that tau and an indirect marker of arterial stiffening (pulse pressure) may synergistically contribute to memory decline, whereas Aβ may have a lesser role in predicting cognitive progression. Tau and vascular pathology (particularly in combination) may represent valuable targets for interventions intended to slow cognitive decline.
INTRODUCTION
The recent FDA approval of aducanumab, which purports to slow the progression of Alzheimer’s disease (AD) through anti-amyloid mechanisms, has furthered the supposition that amyloid-β (Aβ) is the primary catalyst of cognitive and clinical decline in AD [1–3]. However, the questionable efficacy of aducanumab in ameliorating cognitive decline despite its successful targeting and reductions of Aβ [1, 5], coupled with other negative anti-Aβ clinical trials [6] and research demonstrating little to no association between Aβ burden and cognitive outcomes [7–9], suggests the need to identify alternative pathologic targets in AD treatment trials.
These alternative pathologic targets would ideally be informed by evidence for their association with early cognitive decline in a prodromal stage of AD, such that they could be targeted for treatment prior to overt dementia. Accordingly, tau has been identified as a promising intervention target with extensive research demonstrating its critical role in promoting the characteristic progression of cognitive decline observed in AD [10–13]. Specifically, as tau pathology advances in its stereotypical spatiotemporal pattern, there are concomitant changes in cognitive domains subserved by these brain regions susceptible to the neurodegenerative effects of tau [14, 15]. Notably, this well-documented association between tau and cognition can occur independently of Aβ, although the association may be strengthened by concurrent Aβ pathology [8, 10]. Indeed, our prior study demonstrated that a large proportion of older adults exhibit elevated medial temporal tau positron emission tomography (PET) in the context of Aβ PET negative status, and that these individuals demonstrated subtle cognitive compromises greater than that observed in the pathologically normal (i.e., A–/T–) group [16].
Beyond the role of these traditional AD pathologic markers in disease progression, emerging research suggests that vascular risk factors and cerebrovascular pathology also influence AD-related cognitive decline [17, 18]. Although cerebrovascular pathology can be identified through brain-based measures, peripheral assessment of cardiovascular risk provides a reliable, low-cost, and accessible method to index the potential for cerebrovascular insult. Hypertension has been associated with AD risk, and interventions aimed at reducing high blood pressure have demonstrated a reduction in risk for mild cognitive impairment [19–21]. Pulse pressure, which reflects arterial stiffening, can be easily obtained from standard blood pressure measurements and has been associated with elevated baseline and longitudinal cerebrospinal fluid tau levels, as well as memory decline and more rapid progression to dementia [22–24]. Indeed, it has been demonstrated that pulse pressure predicts cerebrovascular disease in the context of AD-confirmed pathology, whereas standard blood pressure measurements (i.e., systolic or diastolic blood pressure) were not predictive [25]. Thus, there may exist an important synergistic effect between cardiovascular risk and tau pathology such that tau-related cognitive decline is exacerbated in the presence of elevated cardiovascular risk, similar to evidence that tau-related cognitive decline is exacerbated among apolipoprotein (APOE) ɛ4 carriers [9]. Notably, this effect may be bidirectional such that cerebrovascular pathology is accelerated by the presence of tau pathology. Regardless of directionality, this interactive effect may have implications for the development of novel AD treatment regimens that simultaneously target tau and cerebrovascular pathology through a combination of anti-tau therapies and intensive blood pressure control.
Therefore, we used PET imaging and sensitive neuropsychological measures to examine the moderating effect of pulse pressure on associations between 1) tau and multi-domain cognition independently of Aβ and 2) Aβ and multi-domain cognition independently of tau. Based on existing literature that demonstrates a robust relationship between tau and cognition as well as more recent evidence for an effect of pulse pressure on biomarkers of tau and dementia risk, we predicted a significant interactive effect between pulse pressure and tau PET such that the negative association between tau and cognition would be strengthened as a function of increasing pulse pressure. We expected the strongest effect with memory and executive function domains given the particular susceptibility of memory with AD pathology and the particular susceptibility of executive function with cerebrovascular pathology [26].
MATERIALS AND METHODS
Standard protocol approvals, registrations, and patient consents
This study was approved locally by the University of California San Diego Human Research Protections Program (protocol #190618), which can be reached at 858-246-4777. Written informed consent was waived for this retrospective data analysis.
Study data
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (https://adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD. For up-to-date information, see https://www.adni-info.org.
Participants
This study included 139 older adults from ADNI who had concurrent tau PET, amyloid PET, blood pressure data, and baseline and 1-year follow-up neuropsychological testing. This sample is predominately White (90.6%) and highly educated (mean 16.44 years; standard deviation [sd] 2.50 years). The mean (sd) age of this sample is 73.87 (7.53) years, with 51.8% of participants being female. At baseline, 68.3% of the sample were cognitively unimpaired (CU) and 31.7% had MCI based on comprehensive neuropsychological criteria [27]. A full breakdown of descriptive statistics for demographic variables, biomarker variables, and cognitive variables split by cognitive diagnosis are found in Table 1.
Descriptive statistics for demographic, biomarker, and baseline cognitive variables split by cognitive diagnosis. All data reflect untransformed values
CU, cognitively unimpaired; MCI, mild cognitive impairment; N, sample size; SD, standard deviation.
Biomarker variables
Cardiovascular risk was indexed using baseline pulse pressure, which is a proxy measure for arterial stiffening [25]. Pulse pressure was defined using the following formula, where BP indicates blood pressure: (systolic BP –diastolic BP)/systolic BP. Additionally, Hachinski Ischemic Score (HIS), a composite of vascular risk factors, was included in statistical models to account for the effects of arterial stiffening independent of generalized vascular risk [28]. Finally, participant use of antihypertensive medications (present or absent) at baseline was included in statistical models to account for the effects of medication on pulse pressure values.
PET imaging was used to assess biomarkers of Aβ (Florbetapir or Florbetaben) and tau (Flortaucipir). For Aβ PET, a cortical composite measure region of interest (ROI) was used that included regions vulnerable to early Aβ deposition [29]. For tau PET, a composite meta-temporal ROI was used that included regions representative of mild-moderate tau pathology (i.e., amygdala, entorhinal cortex, fusiform gyrus, inferior temporal gyrus, and middle temporal gyrus) [30]. Standardized uptake variable ratios (SUVRs) were calculated by dividing the SUV for each ROI by the whole cerebellum SUV (Aβ PET) or the inferior cerebellar gray (tau PET). Aβ SUVR values were converted to a centiloid scale to standardize across the two PET tracers [29].
Cognitive variables
Composite scores for memory, language, and attention/executive function domains were calculated using the following neuropsychological measures: Auditory Verbal Learning Test delayed recall and Logical Memory delayed recall (memory); animal fluency and the Boston Naming Test (BNT)/Multilingual Naming Test (MiNT; language); and the Trail Making Test Parts A & B (attention/executive function). Note that participants either had the BNT or the MiNT as a measure of naming; these scores were converted to percent correct to place them on the same scale and create one single “naming” measure. Z-scores were calculated for individual neuropsychological measures using predicted values relative to an ADNI robust normal control group with available neuropsychological data (i.e., remained cognitively intact throughout the duration of their participation, n = 525) that adjusted for age, sex, and education level. Cognitive domain scores at baseline and year 1 were included to assess longitudinal performance.
Statistical analysis
All biomarker and cognitive variables were transformed using Box-Cox transformation to improve normality and reduce the influence of outliers, which are reflected in the unstandardized regression coefficients. Age, sex, cognitive classification (i.e., CU or MCI), presence of hypertensive medications, and HIS score were adjusted for in all analyses. The first set of models assessed the 1) interactive effect between pulse pressure and tau PET on 1-year cognitive outcomes while adjusting for Aβ PET, demographic covariates, and baseline cognitive performance for a given domain, or 2) interactive effect between pulse pressure and Aβ PET on 1-year cognitive outcomes while adjusting for tau PET, demographic covariates, and baseline cognitive performance for a given domain. These same models were examined with baseline cognition as the outcome variable. The second set of models examined the main effects of pulse pressure, tau PET, and Aβ PET on year 1 cognitive outcomes.
RESULTS
After adjusting for all covariates, there was a significant interaction between pulse pressure and tau PET on 1-year memory performance such that the combination of higher pulse pressure and higher tau PET was associated with lower memory at follow-up (B = –1.76, 95% CI = [–3.42,–.09], t = –2.08, p = 0.04, partial η2 = 0.03; see Fig. 1). This effect was not significant for baseline memory performance (B = 0.18, 95% CI = [–2.49,2.85], t = 0.14, p = 0.89, partial η2 < 0.001). There was not a significant interaction between pulse pressure and tau PET on baseline (B = 668.70, 95% CI = [–894.41,2231.80], t = 0.85, p = 0.40, partial η2 = 0.004) or 1-year (B =–444.23, 95% CI = [–1294.10,405.63], t = –1.03, p = 0.30, partial η2 = 0.008) language performance. There was not a significant interaction between pulse pressure and tau PET on baseline (B =4004.98, 95% CI = [–2058.97,100068.93], t = 1.30, p = 0.19, partial η2 = 0.009) or 1-year (B = 319.90, 95% CI = [–432.55,1072.33], t = 0.84, p = 0.40, partial η2 = 0.005) attention/executive function performance.
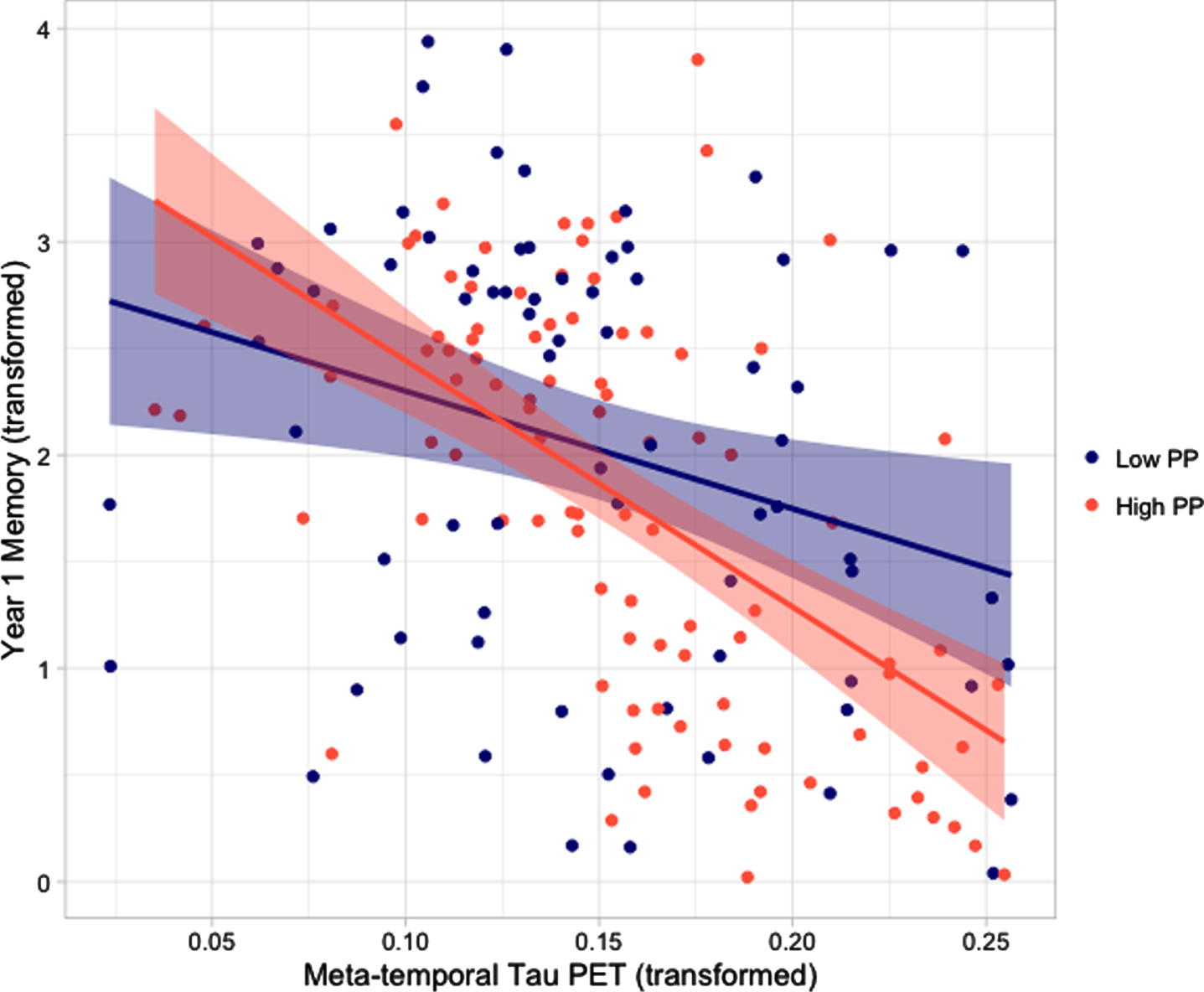
Scatterplots depicting the association between meta-temporal tau PET and memory performance at year 1 for participants with high (red) and low (navy) pulse pressure. Pulse pressure categories were determined by median split. All variables have undergone Box-Cox transformation to improve normality. PET, positron emission tomography; PP, pulse pressure.
After adjusting for all covariates, there were no significant interactions between pulse pressure and Aβ PET on 1-year memory (B = –0.07, 95% CI = [–0.16,0.01], t = –1.68, p = 0.10, partial η2 = 0.02), language (B = –22.73, 95% CI = [–65.75,20.28], t = –1.05, p = 0.30, partial η2 = 0.009), or attention/executive function (B = 2.26, 95% CI = [–35.70,40.23], t = 0.12, p = 0.91, partial η2 < 0.001) performance. Additionally, there was no significant interactive effect on baseline memory (B = 0.07, 95% CI = [–0.07,0.22], t = 0.98, p = 0.33, partial η2 = 0.006), language (B = 20.5, 95% CI = [–64.09,105.11], t = 0.48, p = 0.63, partial η2 = 0.002), or attention/executive function (B = 166.41, 95% CI = [–162.13,494.96], t = 1.00, p = 0.32, partial η2 = 0.006) performance.
After adjusting for covariates including pulse pressure and Aβ PET, tau PET had a significant main effect on 1-year memory performance (B = –2.67, 95% CI = [–4.94,–0.41], t = –2.33, p = 0.02, partial η2 = 0.04), but not language (B = –795.42, 95% CI = [–2013.15,422.30], t = –1.29, p = 0.20, partial η2 = 0.01) or attention/executive function performance (B = –778.10, 95% CI = [–1817.29,261.12], t = –1.48, p = 0.14, partial η2 = 0.02). After adjusting for covariates including pulse pressure and tau PET, there was no significant main effect of Aβ PET on 1-year memory (B = –0.002, 95% CI = [–0.12,0.11], t = –0.04, p = 0.97, partial η2 = < 0.001), language (B = 2.67, 95% CI = [–58.22,63.56], t = –0.09, p =0.93, partial η2 < 0.001), or attention/executive function performance (B = –23.28, 95% CI = [–74.11,27.56], t = –0.91, p = 0.37, partial η2 = 0.006). After adjusting for all covariates including tau PET and Aβ PET, there was no significant main effect of pulse pressure on 1-year memory (B = –0.02, 95% CI =, t = –0.50, p = 0.62, partial η2 = 0.002), language (B = 5.43, 95% CI = [–39.08,49.95], t =–0.24, p = 0.81, partial η2 < 0.001), or attention/executive function performance (B = –17.61, 95% CI = [–55.46,20.24], t = –0.92, p = 0.36, partial η2 =0.007).
DISCUSSION
Our findings indicated that pulse pressure significantly interacts with tau PET to predict 1-year memory. Specifically, those with higher pulse pressure demonstrated a stronger negative association between baseline meta-temporal tau PET and 1-year memory after adjusting for demographic factors, antihypertensive medication use, HIS score, baseline memory, and baseline Aβ PET.
Many studies have demonstrated a robust association between markers of tau pathology, including tau PET, and cognition across multiple domains [9, 13]. Our study expands upon this literature to show that this association is exacerbated in the presence of cardiovascular risk as indexed by high pulse pressure. This synergistic effect of tau pathology and cardiovascular risk on cognition can be explained by examining the pathophysiological effects of cerebrovascular insults, which are elevated in the context of higher cardiovascular risk [31, 32]. Cerebrovascular insults including damage to the blood-brain barrier (BBB), which has been shown to be directly impacted by arterial stiffness-related widening of tight junctions [33], and cerebral hypoperfusion may exacerbate the negative effects of tau through several mechanisms. For one, neurovascular uncoupling and associated cerebral hypoperfusion may induce neuronal vulnerability that increases susceptibility to the pathologic effects of tau neurofibrillary tangles [34, 35]. Another possible mechanism involves injury to the tight junctions of the blood-based barrier that results in inflammatory cytokine activation, which in turn leads to increased tau phosphorylation and subsequent neurofibrillary tangle-related cognitive decline [36–38]. Indeed, one study investigating the relation of vascular markers with tau PET found that both cerebral blood flow and a cerebrospinal fluid marker of pericyte injury were associated with tau, and that tau mediated associations between these vascular markers and global cognition [39]. Interestingly, there appears to be a bidirectional relationship between cerebrovascular pathology and tau pathology such that the latter can also induce vascular injury, and this reciprocal influence appears to be strongly related to neuroinflammatory processes [36, 40]. Future studies are needed to examine the complex relationship between vascular risk, neuroinflammation, and tau pathology, as well as their independent and interactive effects on cognition.
Prior research has demonstrated that tau exerts negative effects on multi-domain cognition after adjusting for Aβ, suggesting that the association between tau and cognition remains significant regardless of the presence and degree of Aβ [9]. Notably, the observed interactive effect of pulse pressure and tau PET on memory in the current study was also evident beyond the main effect of Aβ PET level. These findings support the notion that tau may have an important role in AD pathogenesis beyond the effect of Aβ PET, which could be considered contradictory to existing AD biomarker frameworks that necessitate the presence of Aβ in their characterization of the AD diagnostic continuum [41]. Our results further indicate the need for these influential biomarker frameworks to consider vascular contributions to the AD prodrome, given that the combination of high tau burden and elevated pulse pressure was most strongly associated with memory performance in our sample.
Despite the occurrence of an interactive effect of tau PET and pulse pressure independently of Aβ, we cannot rule out the possible contribution of Aβ to this dynamic interplay of risk factors, and there may exist an additional unique mechanism by which vascular risk interacts with Aβ. Such an effect was observed in a study demonstrating an interactive effect of Aβ PET and a composite measure of cardiovascular disease risk on global cognitive decline, although the effect was not examined independently of tau PET, which may have explained some of the variance in the outcome [42]. Interestingly, another study examining the interaction between vascular risk and Aβ found that there was a synergistic effect on future tau PET levels [43]. Other research has demonstrated that associations between cerebral blood flow/pericyte injury and tau PET are strengthened in the presence of Aβ [39]. Consideration of our findings in the context of existing literature suggests that there may be a more complicated relationship between Aβ PET, tau PET, and pulse pressure than characterized by our study alone.
Importantly, our study was conducted in a predominately White, highly educated, and healthy sample. Diversity in aging research samples, particularly racial/ethnic diversity, is crucial given different rates of AD and pathologic profiles [44, 45]. Social determinants of health such as exposure to discrimination, financial instability, and healthcare access likely have a very important influence on these relationships between AD pathology, cardiovascular risk, and cognition [46]. These potential factors could not be assessed in the current study due to the nature of the sample and limitations on the data collected. Before the findings from our study can be used as evidence for the investigation of tau and vascular risk as alternative treatment targets, results must be replicated in a more representative cohort.
An additional limitation of our study included use of pulse pressure as an indirect measure of arterial stiffening. Although more direct measures of cerebrovascular pathology increase the certainty that we are measuring the intended construct, use of pulse pressure as a proxy has more applicability in clinical settings to identify individuals at risk who may benefit from intervention. Other peripheral metrics such as blood pressure variability have also been linked to AD-related cognitive impairment and may provide additional insight into the moderating role of vascular risk on tau-associated cognitive decline [47]. Strengths of this study include use of sensitive neuropsychological measures across cognitive domains, assessment of longitudinal cognition accounting for baseline performance, adjustment for antihypertensive medication use and general vascular risk, and concurrent analysis of Aβ and tau PET.
Investigation of novel treatment targets are critical for advancement of efforts to slow or stop the progression of AD. As controversy continues around anti-Aβ therapies and their clinical benefits, AD clinical trials are at a critical juncture with an opportunity to shift focus away from Aβ and pursue alternate pathways. Our findings suggest that tau pathology and vascular risk represent viable targets for intervention that have a direct impact on cognition. Indeed, a recent intervention trial determined that intensive blood pressure control was effective at slowing cognitive decline [48], and investigations of anti-tau therapies are currently underway [49]. However, our demonstration of an interactive effect between tau and pulse pressure suggests that a multipronged therapeutic approach that simultaneously intervenes on these targets may be particularly effective in slowing cognitive decline.
Footnotes
ACKNOWLEDGMENTS
Funding for this study was provided by grants from the National Institute on Aging (R01 AG049810 and R01 AG054049 to Dr. Mark Bondi) and the National Science Foundation (DGE-1650012 to Alexandra Weigand). We thank the investigators and participants of ADNI for the use of these data.
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (![]() ). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.