
Abstract
Background:
Numerous studies have shown that the complement system plays an important role in Alzheimer’s disease (AD). However, whether complement 4 (C4) protein in cerebrospinal fluid (CSF) was associated with AD pathology, especially in the early stage of AD, is still unclear.
Objective:
We aimed to explore the association of CSF C4 with AD pathology and cognition in the preclinical AD.
Methods:
The study included a total of 287 participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. Based on the A/T scheme, they were divided into four groups to access the changes of CSF C4 in the preclinical AD. Linear regression models were used to test the associations between CSF C4 and AD core biomarkers, namely Aβ42, P-tau, and T-tau.
Results:
The level of CSF C4 decreased in the A + T- group compared with the A-T- group (p = 0.04) and it increased in the A-T+ group compared to the A + T- group (p = 0.01). In pooled samples, C4 was significantly associated with AD core biomarkers (all p < 0.05), but only in the A + group after stratification according to the A/T scheme. Furthermore, CSF C4 levels at baseline were associated with longitudinal cognitive changes.
Conclusions:
Our results showed that CSF C4 levels changed dynamically in the preclinical AD, and that the responses of CSF C4 to brain Aβ pathology, tau pathology and neurodegeneration were found only in the presence of amyloid plaques, both of which indicates the complex link between C4 and AD.
INTRODUCTION
Alzheimer’s disease (AD) is a common neurodegenerative disease characterized by pathological hallmarks including amyloid-β (Aβ) protein deposits and neurofibrillary tangles composed of hyperphosphorylated tau (p-tau) proteins, followed by prototypical clinical impairment [1]. Aberrant synapse function and aberrant elimination is a key pathomechanism for cognitive impairment in AD [2–5]. The complement system, an integral part of the innate immune system, can rapidly recognize and eliminate pathogens and cellular debris in the brain, which play a major role in the normal function of synapse. Lots of studies demonstrated that the complement system was also involved in physiology and pathology of AD. As shown in AD mouse models, complement and microglia induced the loss of synapses in the early stage of AD [6–8]. Furthermore, activated complement products, a driver of neuroinflammation, is involved in recruiting and activating microglia at sites of fibrillar Aβ deposition in AD [9, 10].
C4 protein is a critical component of the classical complement cascade, functionally involved in synapse clearance during developmental maturation of a neuronal circuit [11]. There are two isoforms of complement C4, namely C4A and C4B. C4A and C4B have different hemolytic activities, covalent affinities for antigens and immune complexes, although they differ by only four amino acids [12]. These differences infer that C4A may be functionally advantageous in ensuring antibody-antigen deposition resolution, whereas C4B has a greater role in propagating the activation pathway leading to membrane attack complex formation when foreign antigens are attacked [11–15]. Furthermore, previous studies have reported the associations of C4 gene expression with multiple neurodegenerative disorders [16, 17]. Massively parallel reporter assays showed that C4A was a risk gene in AD [18] and a previous analysis of UK Biobank data demonstrated that C4A was associated with cognitive performance and brain atrophy [19]. However, the role of C4 protein in the pathogenesis of AD is poorly understood, particularly in the early stages of the disease.
In this study, we investigated CSF C4 levels in different pathological stages of AD and tested the associations of CSF C4 with Aβ and tau pathologies. We also explored whether C4 mediated the effects of Aβ pathology on tau pathology and the predictive values of C4 in cognitive decline. To achieve these aims, the biomarker-based A/T classification system, which has been developed and refined by the National Institute on Aging and the Alzheimer’s Association (NIA-AA), was used to explore these associations since changes in CSF Aβ42 and P-tau levels represent the pathological progression of AD. This classification system consists of two biomarker dimensions including the assessment of Aβ pathology (A) and tau pathology (T) and divided the stage of preclinical AD into 3 successive stages. All participants were non-demented individuals, as non-dementia represents the early stages of AD, which allowed us to study the association between the complement pathway and AD pathology at a very early stage of AD.
METHODS
Subjects
The Alzheimer’s Disease Neuroimaging Initiative (ADNI) database is a research project investigating the relationships between clinical, cognitive, imaging, genetic, and biochemical biomarker features across the spectrum of AD in people with dementia, those with mild cognitive impairment (MCI), and controls at risk of developing cognitive decline and dementia [20]. Exclusion criteria of ADNI database included inability to speak English or Spanish, inadequate visual and auditory capacities for neuropsychologic assessment, central nervous system infection, recent head trauma, active substance abuse, medical contraindication for magnetic resonance imaging (MRI), currently being enrolled in other studies, and poor general health with diseases precluding enrollment [21]. The project was approved by the Emory University Institutional Review Board, and informed consent was obtained from all subjects or their authorized representatives.
There were 134 participants with MCI and 84 healthy controls (HCs) with available CSF C4 data selected from the ADNI database. Participants with no CSF biomarker data and those with data outside three standard deviations (SDs) were excluded. To study the early phase of AD, we also excluded demented participants. Finally, our study included 218 subjects with APOE4 genotyping, baseline measurements of CSF C4 and AD core biomarkers as well as at least one clinical follow-up assessment.
Cognitive assessments
We obtained performance measures on five cognitive tests from the ADNI, including the Mini-Mental State Examination (MMSE) test, the Alzheimer Disease Assessment Scale (ADAS) test, the Preclinical Alzheimer’s Cognitive Composite (PACC) test, the Functional Assessment Questionnaire (FAQ) test, and the Clinical Dementia Rating (CDR) test. The PACC test can track the earliest cognitive changes associated with underlying AD pathology [22]. The ADAS was designed to assess the severity of cognitive and non-cognitive dysfunction in AD patients [23]. Additionally, the FAQ test is a reliable and stable measure of activities of daily living for use in clinical or research settings [24]. The CDR is the gold standard for the staging of dementia due to Alzheimer’s disease [25, 26]. Notably, higher values for the ADAS13, FAQ and CDR and lower values for the PACC indicate poorer cognitive performance.
Measurements of CSF biomarkers
In the ADNI database, CSF samples were collected in the morning after an overnight fast at the baseline visit and then were frozen within 60 min once collected. CSF samples were processed, aliquoted, and stored at –80°C according to ADNI Biomarker Core Laboratory Standard Operating Procedures [27–29]. Aβ42, T-tau, and P-tau were measured by using the Elecsys β-amyloid CSF, the Elecsys phosphotau CSF, and Elecsys total-tau CSF immunoassays on a cobas e601 analyzer (software version 05.02) according to manufacturer’s provisional kit instructions [30]. A peptide sequence called GSFEFPVGDAVSK is located in the alpha chain of C4 (MG7 domain), mainly used for C4 quantification by the multiple-reaction monitoring (MRM) methodology. The raw data and all the intermediate steps are available online at adni.loni.ucla.edu under the Biomarkers Consortium CSF Proteomics MRM dataset [31].
The A/T classification
The A/T scheme, using specific cut-offs for pathological levels of Aβ and tau isoforms, was used to assign ADNI participants to different biomarker groups. The A/T scheme is comprised of two groups of biomarkers: “A” aggregated amyloid pathology (as indicated by CSF Aβ42) and “T” aggregated tau (as indicated by CSF P-tau), as previously reported [32]. And each biomarker group is binarized as either negative (-) or positive (+). “A+” participants refer to those with CSF Aβ42 < 976.6 pg/ml and “T+” participants refer to those with CSF P-tau >21.8 pg/ml [33]. There were then four different biomarker groups combined, namely 1) A-T-, 2) A + T-, 3) A + T+, and 4) A-T+ [32]. We did not apply the full A/T/N scheme as P-tau and T-tau were highly correlated. Furthermore, participants can be classified into the healthy control group (A-T-), the AD continuum group (A + T- and A + T+), and the suspected non-AD pathology group (A-T+) [34–36].
Statistical analyses
Before the analyses, all continuous variables used in this study were log-transformed to normalize if the Kolmogorov-Smirnov test implied they were not normally distributed. Baseline AD core biomarker outliers are defined as three standard deviations (SD) above or below the population mean. Age, sex, APOE ɛ4 carrier status, and education level were adjusted as covariates in all analyses.
First, one-way analysis of covariance (ANCOVA) was performed to determine whether CSF C4 levels differed between the four A/T groups, followed by Bonferroni-corrected post hoc pairwise comparisons. Next, multiple linear regression models were performed separately in the healthy controls, the AD continuum, and the SNAP groups to assess associations between CSF C4 and AD core biomarkers. These analyses were performed in all participants and then in subgroups stratified by diagnosis including the HCs and MCI groups. Furthermore, we investigated whether CSF C4 could be a modulator of the association between amyloid pathology and tau pathology. To achieve this, we conducted casual mediation analyses using linear regression models fitted according to the methods proposed by Baron and Kenny, with CSF C4 as the mediator [37]. In this model, CSF Aβ42 levels were the independent variable, and CSF P-tau levels were the dependent variable. In addition, linear mixed effects (LME) models were used to examine the longitudinal effects of CSF C4 on changes in cognitive function. The LME model has a random time intercept and slope and an unstructured random effects covariance matrix, with time (continuous) and dependent variable (cognitive function) interactions as predictors. The analyses described above were carried out using R (version 3.5.1) and IBM SPSS Statistics 23. A two-sided p-value <0.05 was used for all statistical significance in this study.
RESULTS
Baseline participants characteristics
The demographic features of each group were shown in Table 1. Our sample of 218 participants included 85 (39.0%) women and 133 (61.0%) men. Among all the participants, 84 (38.5%) were cognitively normal (CN) and 134 (61.5%) were diagnosed with MCI. The total participants had an average age of 75.0 and average education of 15.9 years. There were differences in APOE ɛ4 carrying status, biomarkers, and cognitive assessment results between A/T groups (all p < 0.001). However, no differences in age, gender, or level of education were observed between the four groups.
Characteristics of participants by biomarker framework
SNAP, Suspected non-Alzheimer disease pathology; F, female; M, male; APOE, Apolipoprotein E; CSF, cerebrospinal fluid; MMSE, Mini-Mental State Examination; ADAS, Alzheimer’s Disease Assessment Scale; CDR, Clinical Dementia Rating; FAQ, Functional Activities Questionnaire, PACC, Preclinical Alzheimer Cognitive Composite.
CSF C4 levels in different biological stages of AD
To assess the associations of CSF C4 levels with Aβ deposition and the downstream processes of tau pathology and neurodegeneration, we compared C4 levels among different subgroups stratified by A/T scheme, including A-T- group (n = 53), A + T- group (n = 34), A + T+ group (n = 99), and A-T+group (n = 32). As shown in Fig. 1, there were significant differences in the level of CSF between four biological groups (p < 0.001), and both the A-T- (p = 0.038) and A-T+ (p = 0.005) groups have significantly increased CSF C4 levels compared to the A + T- group.
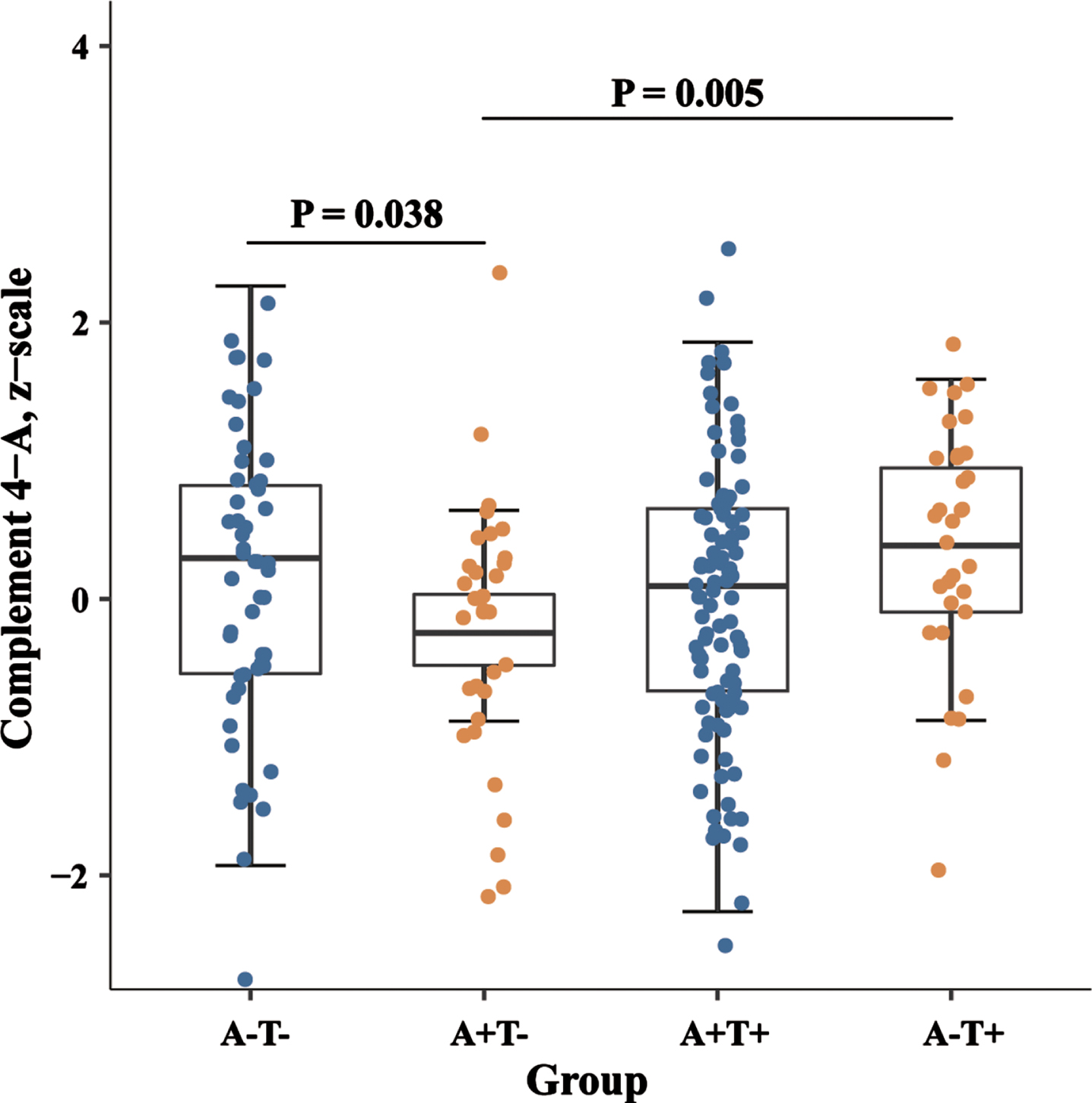
Levels of CSF C4 in the biomarker classification. Boxplots of CSF C4 levels in different stages based on biomarker categories. p-values were assessed by a one-way ANCOVA, and significant p-values after Bonferroni corrected post hoc pairwise comparisons are marked. CSF, cerebrospinal fluid; SNAP, suspected non-Alzheimer disease pathology.
Associations between CSF C4 and AD core biomarkers in AD continuum
We used linear regression models adjusted for age, gender, education level, and APOE ɛ4 carrier status to examine the associations of CSF C4 with AD core biomarkers (Aβ42, P-tau, and T-tau). As shown in Fig. 2, increased CSF C4 was associated with higher levels of CSF Aβ42 (β= 0.284, p < 0.001), CSF P-tau (β= 0.215, p = 0.002), and CSF T-tau (β= 0.274, p < 0.001) in all participants. The results of the subgroup analysis showed that the relationship between CSF C4 and the above CSF biomarkers remained unchanged in the MCI group. However, no significant association was found between CSF C4 and CSF T-tau or P-tau in the HCs group (Supplementary Table 1). After stratification according to A/T scheme, we tested the associations in three groups, the HC group, the AD continuum group, and the SNAP group. Higher levels of CSF C4 were still significantly associated with CSF Aβ42 (β= 0.217, p = 0.008), CSF P-tau (β= 0.309, p < 0.001), and CSF T-tau (β= 0.349, p < 0.001) in AD continuum group, whereas no significant associations were found in the HC or SNAP group.
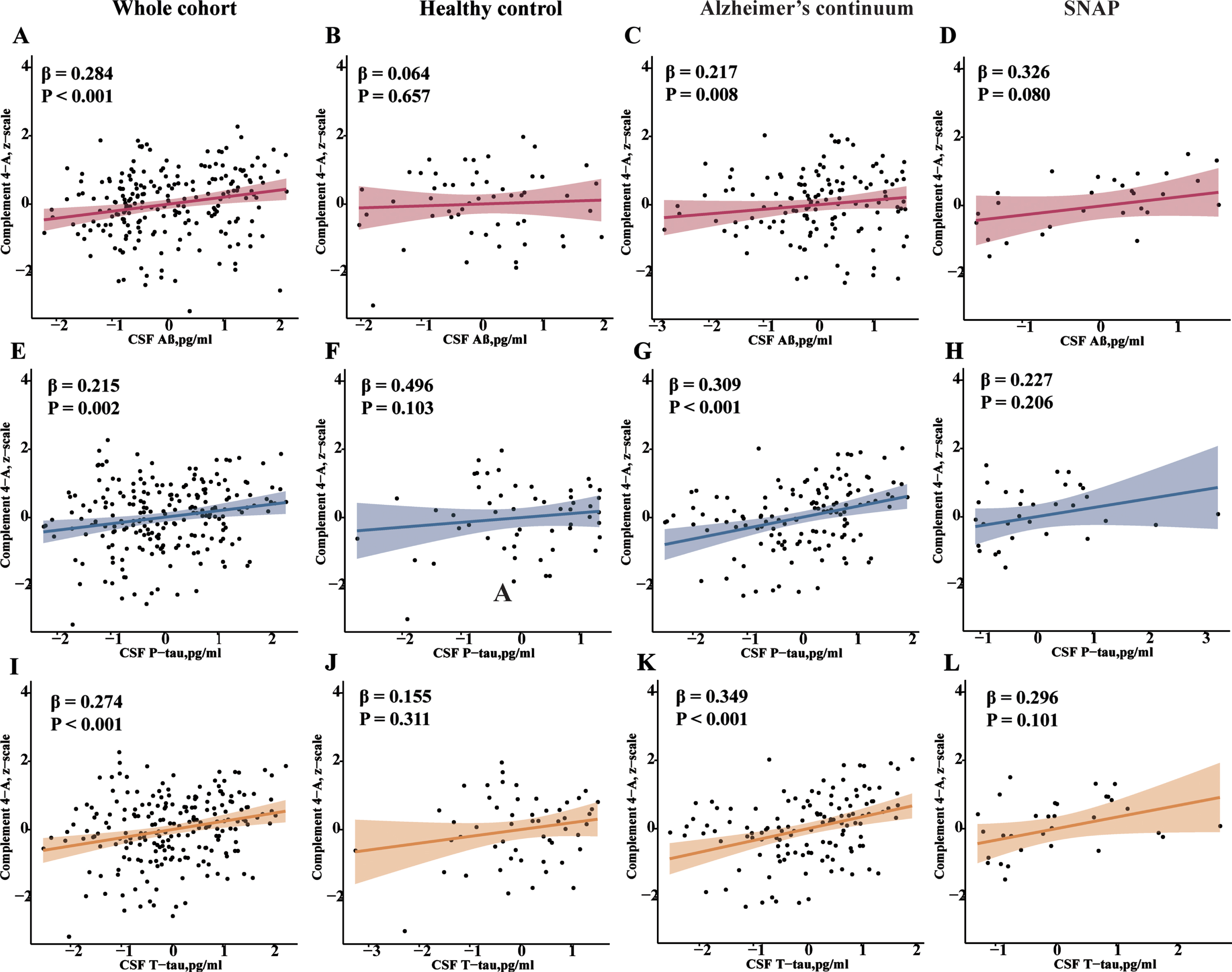
Associations of CSF C4 with AD core biomarkers. Scatterplots show a positive correlation between C4 and AD core biomarkers in the whole cohort. After stratification by the A/T scheme, this association was only significant in the AD continuum group (A + groups). p values were gained via multiple linear regression adjusting for age, gender, educational level, and APOE ɛ4 carrier status. CSF, cerebrospinal fluid; Aβ42, amyloid β42; T-tau, total Tau; P-tau, phosphorylated Tau; APOE, apolipoprotein E.
CSF C4 protein modulated the association between Aβ pathology and tau pathology
To further test the association between C4 and AD pathology, we used mediation analysis to explore whether CSF C4 mediated the association between amyloid pathology (CSF Aβ42) and tau pathology (CSF P-tau). In the total population, CSF C4 was positively associated with CSF Aβ42 and CSF P-tau, while CSF Aβ42 showed a negative association with CSF P-tau, as shown in Fig. 3. Additionally, mediation analyses showed that CSF C4 protein partially mediated the relationship between CSF Aβ42 and CSF P-tau, with a mediating proportion of 39.25%.
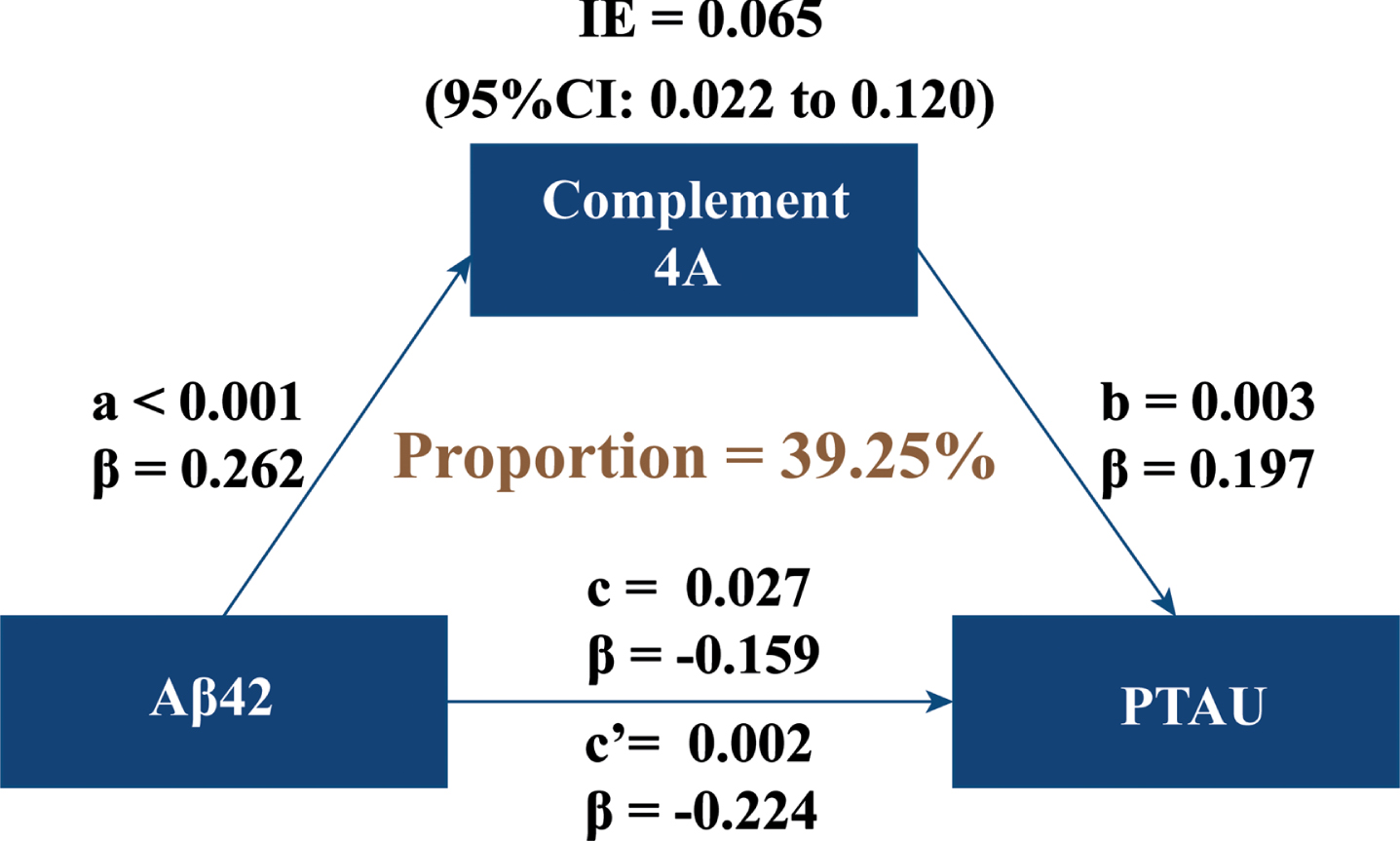
CSF C4 modulated the association of Aβ pathology with tau pathology. Mediation models were created to investigate the mediation effect of C4 on the association between Aβ pathology and tau pathology, with CSF Aβ42 levels as independent variable and CSF C4 levels as mediator and CSF P-tau levels as dependent variable. CSF, cerebrospinal fluid; Aβ42, amyloid-β42; P-tau, phosphorylated tau; IE, indirect effect.
Association between baseline CSF C4 levels and longitudinal cognitive changes
We investigated whether baseline CSF C4 levels were associated with longitudinal cognitive changes in the whole cohort. As shown in Table 2, baseline CSF C4 was negatively associated with ADAS13 scores (p = 0.014) but positively associated with PACC scores (p = 0.043) during the follow-up. Furthermore, CSF C4 at baseline showed no association with longitudinal changes in MMSE, FAQ, or CDR scores.
Associations of CSF C4 with longitudinal cognitive changes
MMSE, Mini-Mental State Examination; ADAS13, Alzheimer Disease Assessment Scale 13; PACC, Preclinical Alzheimer’s Cognitive Composite; FAQ, Functional Assessment Questionnaire; CDR, Clinical Dementia Rating.
DISCUSSION
In the present study, we investigated the changes of CSF C4 in different biological stages of AD and explored the associations of CSF C4 with AD pathologies and cognitive changes in the early phase of AD. Our results showed that CSF C4 levels changed dynamically during the pathological process of AD. Moreover, our results demonstrated that CSF C4 levels were positively associated with CSF Aβ42, P-tau, and T-tau measures in all participants. The results of subgroup analysis by diagnostic classification showed that the relationships between CSF C4 and AD core biomarkers were consistent with the total sample in the MCI subgroup but not HCs subgroup. After stratification by the biological stages of AD, the associations between CSF C4 levels and these biomarkers were still significant in participants within the AD continuum but not healthy control or SNAP group. In addition, our mediation analysis demonstrated that CSF C4 mediated the association between CSF Aβ42 and CSF P-tau. Furthermore, we showed that CSF C4 was positively associated with cognitive performance. Altogether, the above results suggested that C4, like other complement cascade components, was associated with AD pathology, which may affect the subsequent cognitive performance.
The complement cascade plays a very pivotal role in maintaining brain homeostasis. Complements are deficient in the normal brain, but complement levels are significantly increased in the pathological stage of the brain [38]. There is evidence that CSF complement proteins, including CSF C3 and CSF C4, were markedly increased in AD and colocalized into amyloid plaques [39–41]. The dynamic changes in CSF C4 levels at the different pathological stages of AD observed in our study implied that the complement cascade was involved in the early formation of AD pathology. Our results also showed that the A + T- group had the significantly lower levels of CSF C4 compared to the A-T- group, which infers that the concentration of CSF C4 might decrease with Aβ concentration in the onset of AD pathology. In addition, we found that the A-T+ participants had the highest CSF C4 levels among four groups. However, only the difference between A-T+ group and A + T- group was significant, suggesting that complement cascade may also be involved in other neurodegenerative diseases. Our suggestion is supported by a previous study showing that the activation of complement cascade may contribute to synapse loss in neurodegenerative diseases other than AD [10].
Consistent with previous studies, we found that CSF C4 was significantly associated with CSF Aβ42. In our further analysis, this association only existed in A + groups, which suggested that the effect of C4 on amyloid pathology was specific in the pathological stage of AD [42–44]. Furthermore, we showed the significant associations between CSF C4 and CSF tau-related biomarkers (P-tau and T-tau) in total participants, which was in line with a previous study showing that CSF C4 levels were associated with tau deposition as measured by tau PET [45]. After stratification by the A/T scheme, this association was also significant only in A + groups, suggesting that C4 was also involved in the downstream of amyloid pathology in the pathological stage of AD. In addition, the amyloid cascade hypothesis proposed that Aβ deposition in the brain was the initial step in AD pathogenesis, causing tau-immunoreactive neurofibrillary tangles, neuronal loss and finally, clinical dementia [46]. However, based on the previous literature, the role of C4 in the pathological course of AD is less known. Our mediation analysis found that CSF C4 mediated the association between Aβ pathology and tau pathology, indicating that C4 was a potential regulator for the progression of AD pathology.
Additionally, the complement cascade is involved in synapse pruning, which affects cognitive functions [6]. Previous studies also showed that the classic complement pathway related genes and proteins correlated to cognitive functions [19, 48]. In line with these studies, we found that CSF C4 levels was associated with cognitive functions and higher CSF C4 levels predicted better cognitive functions. This finding suggests that C4 as a classical complement component may protect cognitive function by modulating AD pathology, as demonstrated by previous studies that found co-localization of C4 and amyloid plaques, and our study found a strong association between C4 and AD pathology, but further studies are needed to confirm the details between C4 and AD pathology and cognitive function [39, 41].
The underlying mechanisms of effects of C4 on AD pathology remain unclear, several possible explanations are as follows. One possibility is that C4, a component of the complement cascade that binds to the complement component receptor 1 (CR1), may be involved in the classical complement pathway, which can modulate neuroinflammation and neuroimmune response, thereby contributing to the clearance of AD pathology [49, 50]. Another mechanism is that, as shown in a genetic study, a higher expression of C4A is associated with a higher activation of microglia, which can prevent the toxic accumulation of Aβ [7, 51]. Interestingly, a similar explanation has been proposed in several reports, where higher CSF soluble TREM2 (sTREM2), a reported marker of microglial phagocyte activation, is also associated with cognitive decline and reduced AD pathology in non-demented elderly [33, 53]. However, no studies exploring a possible link between CSF C4 and sTREM2 have been identified. The last possible mechanism is that C4 levels were linked to the protective effect of APOE ɛ2 for AD [54, 55]. These mechanistic explanations for the relationship between C4 and AD pathology have yet to be elucidated, and future studies to further elucidate this relationship will be crucial for the development of new treatments to halt the progress of AD pathology.
The strengths of the current study are that it is the first study to systematically investigate the associations between CSF C4 and AD pathology, and that the use of biomarker classification provides the direct information for their associations in different pathological stages of AD. However, our study also has some limitations. Firstly, the limited sample in ADNI may limit the statistical power. Future studies aimed at validating our findings should be carried out in a larger-scale population. Secondly, our analysis of the associations between C4 and AD biomarkers was only performed at baseline, well-designed longitudinal cohorts are needed to verify our findings about the influence of C4 on AD pathologies. Thirdly, the mediation model applied in the current observational studies does not allow us to infer a causal relationship between C4 and AD pathology and cognitive function. Fourthly, we used CSF Aβ42 for amyloid (A) in accordance with the NIA-AA research framework guidelines, whereas a recent study pointed out that Aβ42/Aβ40 and Aβ42/Aβ38 ratios in CSF had better diagnostic accuracy than CSF Aβ42 for detection of brain amyloid deposition in prodromal AD [56]. Thus, our results in this regard should be taken with caution. Fifthly, the C4 data used in our study are log quantified values and are not measured concentrations of the original protein, so the range of differences for this protein in this study cannot be used as a practical clinical reference. The use of reference concentrations of CSF C4 protein in clinical practice should be tested in a larger sample based on its raw concentrations. Finally, the lack of available amyloid and tau PET data prevented us from studying the interaction between C4 and AD at a later stage of AD pathology.
In summary, our study showed that CSF AD core biomarkers were tightly associated with CSF C4 levels at the early stage of AD, and C4 played a key role in the effect of amyloid pathology on tau pathology. Moreover, we also found that higher levels of CSF C4 predicted slower longitudinal cognitive decline. These above findings suggested that C4 could serve as a novel therapeutic target for AD prevention and treatment.
Footnotes
ACKNOWLEDGMENTS
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
FUNDING
This study was supported by grants from the National Natural Science Foundation of China (81971032, 82271475), and Medical Science Research Guidance Plan of Qingdao (2021-WJZD001).
CONFLICT OF INTEREST
Lan Tan is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.