
Abstract
A representative FeN4-doped conical carbon (C) scaffold was investigated for participating in interactions with the thio-substituted fluorouracil (SFU) anticancer drug by performing density functional theory (DFT) calculations. In this regard, all possible relaxation configurations of SFU at the doped tip of C scaffold were examined, in which three models were obtained including one horizontal relaxation configuration (FC1) and two vertical relaxation configurations (FC2 and FC3). The results indicate the highest stability and strength for FC1 model. Examining formations and strengths of interactions showed two medium strength interactions in each of FC1, FC2, and FC3 models. Moreover, the evaluated electronic molecular orbitals features indicated availability of sensor function for the proposed C scaffold towards the interacting SFU substance. As a consequence, the models were determined to work in dual functions of sensor and carrier towards drug delivery purpose of SFU anticancer drug.
Introduction
The innovation of nanotechnology has been always an important issue regarding the expected roles of this novel technology relatives in different applications with especial importance for those of living systems [1–5]. In this regard, developing nanostructures and investigating their mechanism of actions were explored drastically to show the applicability of nanotechnology for biological related fields [6–10]. Besides the early known carbon nanotube, several other types of nanostructures have been also innovated with deterministic features in shapes and compositions [11–15]. Spherical fullerenes, monolayer graphenes, conical nanocones, and several other examples have been included in the category of nanostructures with carbon or non-carbon compositions [16–20]. To this aim, several attempts have been dedicated to recognize features of such novel nanostructures for various purposes [21–23]. Indeed, the field of drug design and development has been always depending on several features to recognize a substance for applications in biological and living systems related fields [24–28]. One of the major expected applications of nanostructures is their role in biological related systems especially for those topics of therapy and medications [29–31]. Earlier works indicated the significant roles of nanostructures for applications in drug delivery systems to increase efficiency of drug impacts on the targeted tissues [32–34]. Several efforts have been dedicated to explore dominant features of the investigated models for assigning their expected functions and applications [35–37]. This is indeed a complicated issue, which should be described very well for providing information in this case [38–40]. To approach such goal, computer-aided drug design (CADD) approach could help very much to provide a small scale system of investigating the related substances of drug delivery systems [41–45]. One of the important issues of knowing possible applications of nanostructures in drug delivery systems is their adsorbing feature of pharmaceutical substances [46–48]. For achieving this goal, several attempts have been dedicated to show interaction details of nanostructures and several other pharmaceutical substances to show the adsorption strength of such system besides evaluating the molecular and atomic scales descriptors [49–51].
Herein this work, we investigated such advantage of nanostructure applications in drug delivery systems by examining the adsorption process of a pharmaceutical substance, thio-substituted fluorouracil (SFU), at the tip of a representative conical carbon (C) scaffold (Fig. 1). As indicated by the achievements of earlier works, doped-models of nanostructures could show dominant features for the investigated systems even better that the original pure nanostructure [52–55]. To show benefits of doing such structural modifications, the tip of C was doped by a combination of FeN4 group resembling the HEME environment (Fig. 1). Hence, the model of C scaffold was prepared for adsorbing the SFU substance to yield SFU@C compounds (GC1, FC2, and FC3) (Fig. 2). Indeed, all possible relaxation configurations of SFU were examined at the FeN4-doped tip region of C, in which three models were obtained. To this aim, performing the quantum chemical density functional theory (DFT) calculations has been seen to be a useful tool for providing information for the investigated models of interaction nano-drug systems [56–60]. As a consequence, such useful tool was employed in this work to provide the required information for discussing the current research goal; examining the SFU adsorption at the tip of conical C scaffold. The obtained quantitative results were listed in Tables 1–3 and those of visual results were exhibited in Figs. 1–3. Fluorouracil is an important pharmaceutical compound with vastly applications in medications of several types of cancers; however, its targeted drug delivery process is still a challenging topic [61–63]. Therefore, several works have been performing to show features of this important anticancer drug in addition to examining hypothesis of proposing novel carriers for pushing the targeted drug delivery processes [64–68]. Within this work, such hypothesis was examined by employing a representative C scaffold for the drug delivery purpose of SFU, which is a thio-substituted derivative of fluorouracil. Earlier works indicated advantages of such thio-substituted derivatives of uracil for working in medications of cancers [69–71].
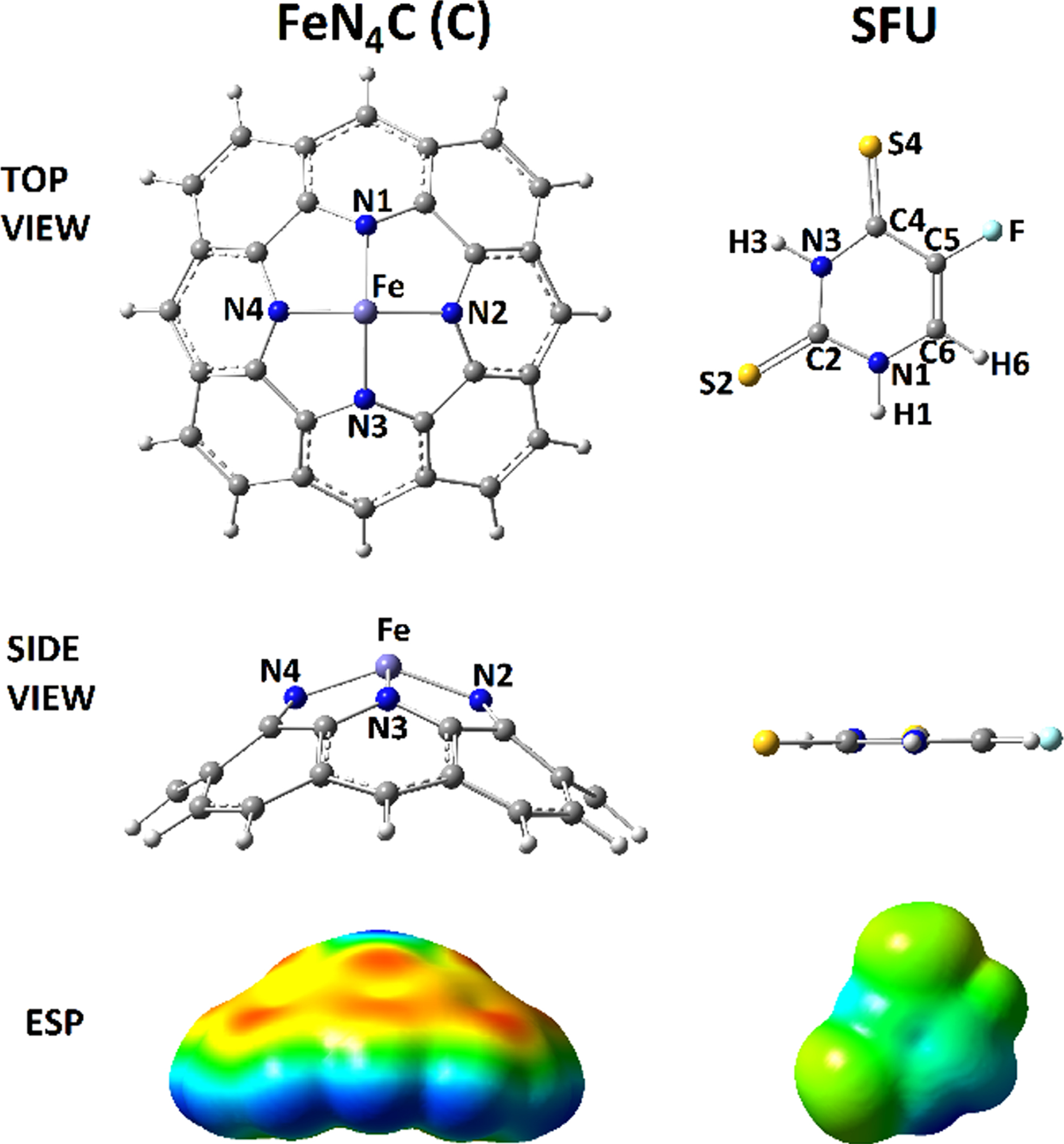
Optimized configurations and ESP surfaces of C and SFU models.
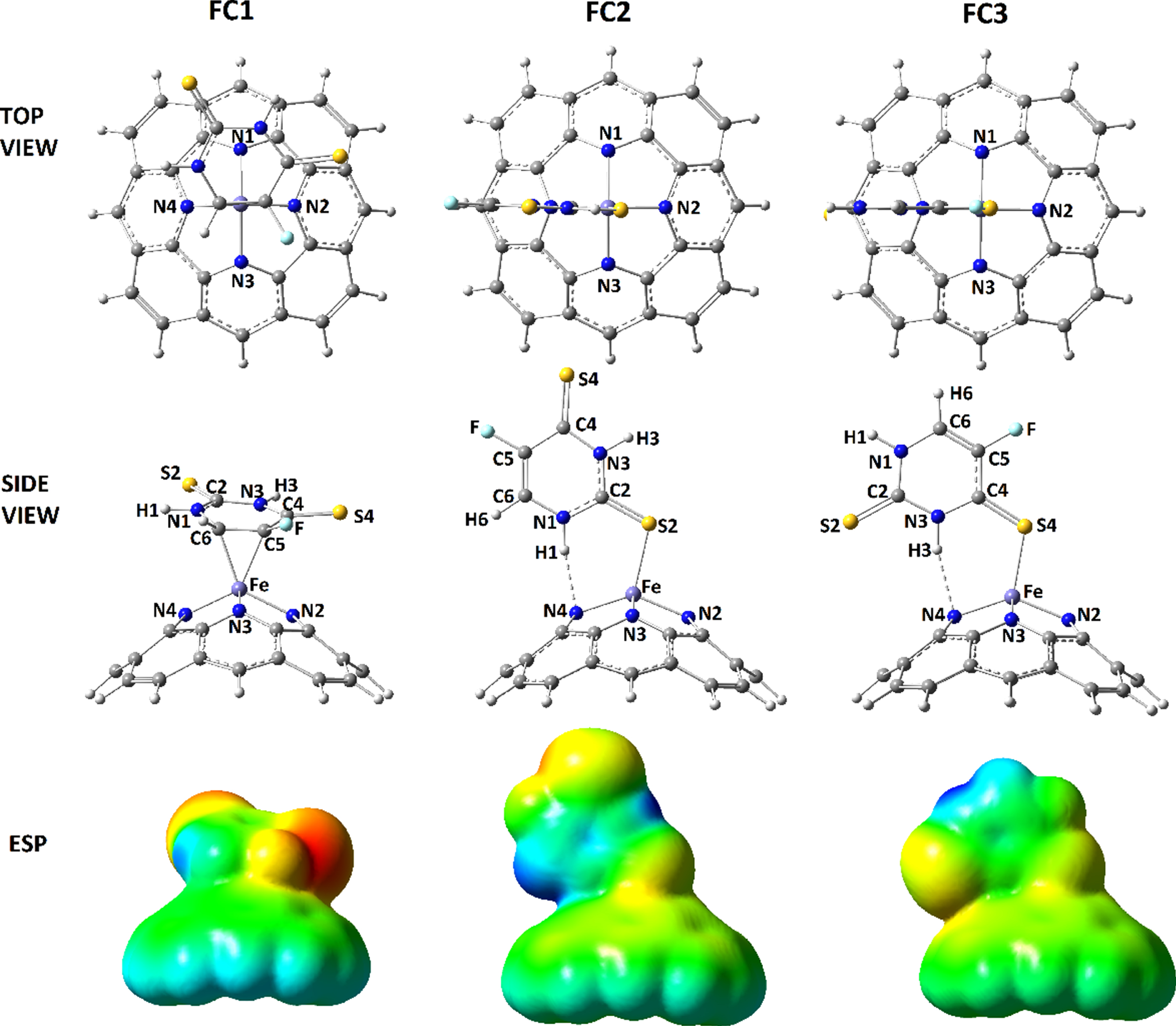
Optimized configurations and ESP surfaces of SFU@C models.
Bond distances of the optimized models
All distances are in angstrom.
Energies and dipole moments of the optimized models
All energies are in electron volt and Dm is in Debye.
Interactions details of the optimized models
Distance is in angstrom and other values are in atomic unit.

HOMO-LUMO distribution patterns and DOS diagrams.
DFT calculations were performed to provide the optimized models of singular FeN4-doped conical carbon (C) scaffold and thio-substituted fluorouracil (SFU) pharmaceutical substance (Fig. 1). The investigated 3D molecular models were drawn and their initial geometries were provided for involving in the optimization calculations to relax to the minimum energy levels. Next, combinations of SFU and C were examined to obtain interacting complexes of the models, in which three models were obtained including FC1, FC2, and FC3, as shown in Fig. 2. Indeed, the FeN4-doped tip of C was targeted for adsorbing the SFU substance, in which all possible relaxation configurations of SFU at the tip of C were examined and the optimization calculations yielded three models of SFU@C bimolecular complexes. Indeed, the obtained structures were those geometries with the lowest possible energy among all energies of the possible structural conformations. Next, the optimized bond distance geometries of those interacting substances were summarized in Table 1. Moreover, different values of energies including total energy (E) and adsorption energy (Eads) were evaluated to recognize the stabilities of models and the energy strength of interacting substances. The value of Eads was obtained by measuring energy differences of complex and singular models to see the extracted energy from the adsorption processes. Electronic molecular orbital energies including energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), energy distances (Eg) of HOMO and LUMO levels, and Fermi energy (Ef) were obtained for the investigated models. The value of Eg was obtained by energy differences of HOMO and LUMO levels to show the energy distance between two major frontier molecular orbitals. The average of HOMO and LUMO values was assigned by the value of Ef. Values of dipole moments (Dm) were also calculated to show electric charge distributions of the investigated models. All these obtained results were summarized in Table 2. Electrostatic potential (ESP) surfaces, HOMO-LUMO distribution patterns, and density of states (DOS) diagrams were also exhibited for the investigated models (Figs. 1–3). To recognize the nature of available interactions between C and SFU substances, the quantum theory of atoms in molecule (QTAIM) [72] was employed to confirm availability and strength of the interactions in two counterparts of FC1, FC2, and FC3 models, and the results were summarized in Table 3. All DFT calculations of this work were performed using the B3LYP/6-31G(d) method and basis set as implemented in the Gaussian program [73]. Based on the achievements of earlier works, performing such computer-aided investigations could help to recognize features of nanostructures, which are not easily achievable in complicated experiments [74–77].
Results and discussion
By the importance of investigating developments of drug delivery systems, this work was performed to show the benefit of employing a representative FeN4-doped conical carbon (C) scaffold for drug delivery of a thio-substituted model of fluorouracil (SFU) anticancer drug. It is important to mention that the atomic doped models of carbon nanostructures could provide an appropriate region for interacting with other substance. Indeed, existence of such FeN4-doped model could provide a HEME-like region, which is well-known for the biological systems as an advantage of decoration of carbon nanostructures for employing in the biological systems. Accordingly, such modified model was targeted for adsorbing the investigated drug structure. To this aim, the singular models (Fig. 1) were prepared for participating in molecular interactions to create bimolecular models of SFU@C as indicated by FC1, FC2, and FC3 (Fig. 2). To approach this step, all possible configurations of SFU at the modified conical tip of C were examined, in which the three mentioned models were obtained. To recognize the impacts of such molecular interactions on geometrical features, the bond distances of singular and bimolecular models were summarized in Table 1. As could be seen for the models, the bond distances were changed from singular to bimolecular models yielding different distances for the models systems. Indeed, in such flexible interacting systems, all bonds are allowed to be re-arranged for reaching to a point of minimized energy configuration. Although this re-arrangement in not avoidable, but knowing the magnitudes of changes are very important for development of formations of such bimolecular systems. Especially in the case of drug delivery systems, it is a must to know variations of structural features of drugs after loading on a nano-surface. The results could show significant variations of bond distances for those bonds close to the interaction regions whereas those of other bonds were remained with few changes. In this case, it could be expected that the energy features should be also changed by such conformational changes. As described in Table 2, both of total energies (E) and adsorption energies (Eads) detected the effects of interactions changes, in which describing the bimolecular models formations could help to recognize details of such changes. In FC1, the initial model of SFU was placed in a horizontal mode at the modified tip of C, in which the SFU model was relaxed through formation of interactions between its C5 and C6 atoms and Fe atom of C. In this case, two direct interactions were occurred between SFU and C. In FC2, the initial model of SFU was placed in a vertical mode with S2 head at the modified tip of C, in which the SFU model was relaxed through formation of interactions between S2 and Fe and between H1 and N4. As a consequence, two interactions were observed for formation of this model with initial placing of S2 head towards the modified tip region. In FC3, the initial model of SFU was placed in a vertical mode with S4 head at the modified tip of C, in which the SFU model was relaxed through formation of interactions between S4 and Fe and between H3 and N4. Accordingly, two interactions were observed for this bimolecular model formation. Based on the obtained values of energies for the formations of bimolecular models, it was observed that the formation stability could be arranged in this order FC1 > FC3 > FC2, in which the values of Eads could also affirm such achievement. Returning to Fig. 1, the exhibited ESP surfaces could show that the tip of C is in blue color and each of S atoms of SFU are in semi-yellow colors. Since the ranges of colors are from negative to positive in this order: Red-Yellow-Green-Light Blue-Blue, the negative sides of SFU were expected to interact with the positive side of C. As a result, the models of bimolecular models of SFU@C were obtained. Interestingly, the colors of those two C5 and C6 carbon atoms of SFU were oriented to yellow, and because of it such dual interaction mode was seen for FC1 model. The light blue color of N-H bond region of SFU helped for formation of such N . . .H interactions between C and SFU in both of FC2 and FC3 models. It is noted that the models were allowed to relax freely, in which three finalized configurations of bimolecular models were obtained for such system based on the calculated values of energies. In this regard, the models were optimized and the hypothesis of bimolecular complex formations of SFU@C was confirmed. However, details of electronic systems should be recognized to help the features of models.
As could be seen in Table 1, the obtained energy values of HOMO and LUMO were changed by bimolecular models formations. The importance of HOMO-LUMO is because of their role in electron transferring processes, in which HOMO stands for the highest occupied molecular orbital and LUMO stands for the lowest unoccupied or vacant molecular orbital. As a result, electron transferring between two levels could lead to electrical conductivity, which is an important issue of sensor function. In this regard, changes of quantities of HOMO-LUMO levels could show impacts of bimolecular formations on such molecular orbital features in addition to showing their role of sensor functions. Moreover, HOMO-LUMO distribution patterns and DOS diagrams of complex models were exhibited in Fig. 3 to show benefit of such adsorption process for sensor function purposes. The energy distance of HOMO and LUMO levels (Eg) could help to show the signaling effect of sensor function, in which such distance was changed from the original C model to each of the bimolecular models of FC1, FC2, and FC3. Accordingly, the Fermi level (Ef) was also changed because of such electronic molecular orbital energy fluctuations. As a consequence, the models showed that the investigated C scaffold could be proposed for drug delivery process of SFU with carrier and sensor dual functions.
Further analyses of the models were done by evaluating interactions details of the models as listed in Table 3. To this aim the interactions distances and QTAIM features including ρ, ∇2 ρ, H were evaluated for examining the formation and strength of interactions. In this regard, signs and magnitudes of the QTAIM features could show the evidence of formation and strength of the interactions. For all three models of SFU@C bimolecular complexes, the interacting substances were placed in reasonable distances towards each other and the QTAIM features were in one negative sign showing reasonable strength of interactions formations. In this regard, if all three ρ, ∇2 ρ, H features were in negative signs, formations of the covalent chemical bonds could be expected. In the case of one negative sign, formations of medium strength interactions could be expected. In the case of SFU@C models, medium strength interactions were observed for the models. Reminding the values of energies, FC1 was at the highest strength, in which the obtained QTAIM features could also affirm such achievement. In more details, values of QTAIM features of FC1 were larger than those of other FC2 and FC3 models. Interestingly, those of FC3 were also larger than those of FC2 revealing higher strength of FC3 than FC2. As a consequence, formations of the interacting models were confirmed and their strength were examined for showing the occurrence of SFU@C complex formations.
Comparing obtained results the investigated models of this work with those results obtained by other works indicated that the current SFU@C models could be expected suitable models for drug delivery purposes [78, 79]. Not only drug delivery, but also drug releasing is an important issue for such important process, in which the physical interactions of SFU and C models could help to release the loaded substance at the correct target. Moreover, the characteristic features of the loaded SFU could be kept close to the original model through a physical interaction. However, in the occurrence of chemical bonds, such features would be changed in an irreversible mode and the drug releasing could not be easily done. As a concluding remark of this work, the investigated SFU@C models could be investigated for further development of such systems of drug delivery purposes.
Conclusion
In this work a representative FeN4-doped conical carbon (C) scaffold was investigated for adsorbing the thio-substituted fluorouracil (SFU) anticancer drug. The optimized calculations indicated availability of three models of SFU@C complexes including FC1, FC2, and FC3. In this case, the strength of FC1 formation was at the highest level with horizontal relaxation at the doped region. Moreover, two other vertical configurations were found to be in the stability order of FC3 > FC2 meaning higher stability of configuration of SFU through S4 atom than S2 atom towards the doped region. In all cases, two medium strength interactions were found between the C scaffold and SFU substance in each of SFU@C bimolecular complexes. Additionally, variations of electronic molecular orbital features indicated availability of sensor applications for such C scaffold. As a consequence, the C scaffold was seen to adsorb the SFU substance with dual functions of sensor and carrier towards approaching the drug delivery purposes.